Medisinsk ekspert av artikkelen

Nye publikasjoner
hamartom
Sist anmeldt: 29.06.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
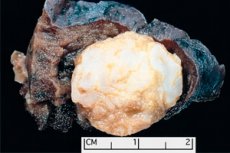
Tumorlignende formasjoner lokalisert i et hvilket som helst anatomisk område som følge av unormal vekst av godartet vev, defineres i medisin som hamartom (fra gresk hamartia - feil, defekt). [ 1 ]
Epidemiologi
Statistisk sett utgjør hamartomer 1,2 % av benigne neoplasmer. Forekomsten av lungehamartomer er anslått til å være omtrent 0,25 % av den generelle befolkningen og står for opptil 8 % av alle lunge-neoplasmer. De fleste lungehamartomer diagnostiseres tilfeldig hos pasienter i alderen 40 til 70 år, men er svært sjeldne i pediatrisk praksis.
Generelt diagnostiseres de fleste hamartomer hos menn, selv om de i nyren er vanligere hos kvinner og identifiseres i middelalderen.
Omtrent 5 % av godartede brystsvulster er hamartomer, og de rammer oftest kvinner over 35 år.
80–90 % av hamartomatøse lesjoner i hjernen og mer enn 50 % av hamartomer i hjertet er assosiert med tuberøs sklerose.
Fører til hamartomer
Gamartomer tilhører medfødte misdannelser og er formasjoner av godartet karakter, som dannes fra mesenkymale vev som stammer fra germinalplater. Og årsakene til deres forekomst er forbundet med ukontrollert celledeling av cytologisk normalt vev (bindevev, glatt muskulatur, fett eller brusk), karakteristisk for en gitt anatomisk plassering, og deres fokale overvekst under embryogenesen av nesten ethvert organ eller anatomisk struktur.
Forekomsten av flere hamartomer hos samme pasient blir ofte referert til som hamartomatose eller pleiotropisk hamartom.
Disse svulstene kan oppstå sporadisk eller i nærvær av visse autosomalt dominante arvelige sykdommer samt genetisk bestemte syndromer.
I mange tilfeller dannes hamartomer når en sjelden genetisk sykdom av multisystemisk natur – tuberøs sklerose – manifesterer seg kort tid etter fødselen, eller ved Recklinghausens familiære sykdom – nevrofibromatose type 1. [ 2 ]
Risikofaktorer
De viktigste risikofaktorene for dannelse av hamartom inkluderer tilstedeværelsen av såkalte genetiske syndromer av hamartomatøs polypose i pasientens historie, inkludert:
- Multippelt hamartomsyndrom - Cowden syndrom, der flere hamartomer av ekto-, ento- og mesodermal opprinnelse dannes, gastrointestinal polypose og mukokutane manifestasjoner observeres;
- Peutz-Jeghers-Turen syndrom (karakterisert ved utvikling av godartede hamartomatøse polypper i mage-tarmkanalen);
- Proteus syndrom;
- Weil syndrom - juvenil polypose i tykktarmen;
- Bannayan-Riley-Ruvalcaba syndrom, som i likhet med Cowden syndrom produserer flere hamartomer (hamartomatøse polypper) i tarmen;
- Carney-Stratakis syndrom og Carney-kompleks.
I tillegg dannes hamartomer hos pasienter med arvelig Watson-syndrom og i tilfeller av sporadisk forekommende eller medfødt Pallister-Hall-syndrom med hypothalamisk hamartom og polydaktyli.
Patogenesen
Mekanismen for økt proliferasjon av germinalvev med dannelse av tumorlignende misdannelser i ulike organer forklares av kromosomavvik og genmutasjoner som kan oppstå spontant eller være arvelige.
Ved tuberøs sklerose er det identifisert mutasjoner i TSC1- eller TSC2-genene – tumorsuppressorer som forhindrer og hemmer overdreven proliferasjon – for rask eller ukontrollert cellevekst og -deling. Og ved nevrofibromatose type 1 og Watson syndrom – kimlinjemutasjoner i det mitokondrielle tumorsuppressorgenet NF1.
Ved hamartom-tumorsyndrom, som kombinerer Cowden-, Protea-, Bannayan-Riley-Ruvalcaba- og juvenil polyposesyndrom, er patogenesen assosiert med mutasjon av PTEN-genet, som koder for et enzym involvert i reguleringen av proliferasjon og regnes som et tumorsuppressorgen.
Mutasjoner i STK11-genet som koder for strukturen og funksjonen til et av de transmembrane serinenzymene, noe som reduserer dets evne til å hemme celledeling, fører til Peutz-Jeghers-Turen syndrom, med utvikling av tarmpolypper og pigmenterte hudlesjoner. En mutasjon i GLI3-genet, en transkripsjonsfaktor involvert i dannelse av intrauterin vev, er identifisert ved Pallister-Hall syndrom.
Dermed fører ukontrollert cellevekst på grunn av genmutasjoner til hamartomdannelse.
Symptomer hamartomer
Avhengig av lokaliseringen av hamartomer, skilles typene deres, og hver av dem har sin egen struktur og symptomatologi.
Hamartom i lungen
Pulmonalt hamartom kan dannes i hvilken som helst lapp og perifere deler av lungene og består av normalt vev som finnes i lungene: fettvev, epitelvev, fibrøst vev og bruskvev. I 80 % av tilfellene dominerer kondroidkomponenten (hyaline bruskceller) med inkludering av adipocytter - fettvevsceller og luftveisepitelceller. [ 3 ]
De tidligere navnene: kondroid hamartom, mesenkym, kondromatøst hamartom eller hamartokondroma anbefales for øyeblikket ikke av WHO.
Mesenkymalt cystisk hamartom i lungen er derimot mindre vanlig og er assosiert med Cowden syndrom hos de fleste pasienter.
Hamartomatøs lesjon i lungen manifesterer seg kanskje ikke, men kan forårsake symptomer i form av kronisk hoste (ofte med hemoptyse), piping i pusten og pustevansker. [ 4 ]
Et hamartom i hjertet
Godartede primære hjertesvulster hos voksne inkluderer modent myocytthamartom, og hos spedbarn og barn med tuberøs sklerose, rabdomyom, det vil si myokardial hamartom i ventriklene eller interventrikulært septum. [ 5 ]
Modent kardiomyocytt-hamartom utvikler seg i ventrikkelveggen (og sjelden i atriene) og kan fremstå som flere lesjoner som er tette masser nært forbundet med det underliggende myokardiet. Svulsten kan forårsake symptomer på hjertesvikt: brystsmerter, hjertebank og arytmier, hjertebilyd, ødem, dyspné, cyanose.
Hjerterabdomyomer, hvorav de fleste diagnostiseres i løpet av det første leveåret, består av hjertemuskelvev dannet av embryonale myoblaster og har utseendet til faste fokale masser uten kapsel.
Vanligvis presenterer disse hamartomene seg asymptomatisk og går spontant tilbake før fylte 4 år.
Hamartomatøse lesjoner anses også av noen eksperter å være assosiert med Carney-kompleksets myxom i hjertet. [ 6 ]
Gamartom i mage-tarmkanalen
Gastrisk hamartom er en mesenkymal masse i form av en epitelial hyperplastisk polypp i magen, Peutz-Jeghers polypp og et sjeldent myoepitelialt hamartom – med hypertrofierte glatte muskelbunter. Andre navn på dette hamartomet inkluderer myoglandulært hamartom, adenomyomatøst hamartom og gastrisk adenomyom. Typiske kliniske manifestasjoner inkluderer dyspepsi, epigastrisk smerte og blødning i øvre mage-tarmkanal. [ 7 ], [ 8 ]
Mer informasjon i materialet - magepolypose
Et intestinalt hamartom er en hamartomatøs eller hyperplastisk polypp i tykktarmen, diagnostisert som et adenomatøst eller tubulært adenom. Når hamartomet er lokalisert i Brunner-kjertelen i tolvfingertarmen, manifesterer symptomene seg ved smerter i epigastriet; kvalme, oppkast og flatulens (som indikerer tarmobstruksjon); og, hvis av betydelig størrelse, gastrointestinal blødning. Ved myoepitelial hamartom i ileum klager pasientene over magesmerter, har redusert kroppsvekt og utvikler kronisk anemi. [ 9 ], [ 10 ]
Les også - rektalpolypper
Retrorektalt hamartom er et cystisk hamartom eller en flerkammercyste i det retrorektale rommet (det løse bindevevet mellom endetarmen og dens egen fascia) som oftest forekommer hos middelaldrende kvinner. Det ser ut som en cyste som buler ut fra den bakre veggen av endetarmen, som er foret med epitel og inneholder kaotisk arrangerte glatte muskelfibre. Dette hamartomet presenterer seg med smerter i nedre del av magen og tilbakevendende forstoppelse. [ 11 ], [ 12 ]
Hamartomer i leveren og milten
Multippelt biliært hamartom i leveren er et hamartom i de interdollice intrahepatiske gallegangene assosiert med misdannelser i utviklingen deres i løpet av embryonalperioden. Dette hamartomet (enkelt eller multippelt) består av tilfeldig utvidede klynger av galleganger og fibrokollagenøst stroma. [ 13 ]
Biliære hamartomer er asymptomatiske og oppdages vanligvis tilfeldig (under radiologisk undersøkelse eller laparotomi). [ 14 ]
En sjelden og ofte tilfeldig oppdaget primær svulst av godartet karakter er et hamartom i milten, som består av elementer av miltens røde pulpa - i form av en veldefinert homogen masse med fast konsistens. Denne misdannelsen kan være enkel eller flerdobbel; når man klemmer miltparenkymet, kan det være en følelse av ubehag og smerte i venstre subkostalområde. [ 15 ], [ 16 ]
Nyrehamartomer
Det vanligste hamartomet i nyren diagnostiseres som angiomyolipom i nyren, ettersom denne godartede svulsten består av modent fettvev med innebygde glatte muskelfibre og blodkar. Den dannes ved tuberøs sklerose i 40–80 % av tilfellene. Økning i størrelsen på hamartomet (mer enn 4–5 cm) fører til smerte og blod i urinen. [ 17 ], [ 18 ]
Hamartom i brystet
De WHO-aksepterte diagnostiske definisjonene av brysthamartom er begreper som adenolipoma, kondrolipoma og myoidhamartom. Selv om mammologer ofte kaller det fibroadenolipoma, fordi tumorformasjonen inneholder celler av fibrøst, kjertel- og fettvev omsluttet av en tynn bindevevskapsel med tydelige konturer. Fokale forkalkninger kan observeres ved visualisering. I dette tilfellet er kliniske manifestasjoner fraværende. [ 19 ], [ 20 ]
Les også - brystsvulster
Hamartomer i hjernen
En tredjedel av pasienter med tuberøs sklerose har et hjernehamartom i form av intrakranielle kortikale utvekster eller tuberkler i forskjellige lapper – ved grensen mellom grå og hvit substans – eller subependymale noduler langs veggene i hjerneventriklene. Astrocytisk hamartom, et subependymalt kjempecelle-astrocytom med kortikal forstyrrelse, dysmorfe nevroner og store gliaceller i hjerneparenkym (astrocytter), kan også dannes. Symptomer på cerebrale hamartomer inkluderer anfall og psykisk utviklingshemming hos barn. [ 21 ], [ 22 ]
En sjelden misdannelse som oppstår under embryogenesen og er tilstede ved fødselen er et hypothalamisk hamartom, som er en masse heterotopiske nevroner og gliaceller. Etter hvert som barnets hjerne vokser, forstørres svulsten, men sprer seg ikke til andre hjerneområder. [ 23 ], [ 24 ]
Hvis det dannes hypertrofiert vev i den fremre delen av hypothalamus (tuber cinereum), der hypofysen fester seg til den, manifesterer misdannelsen symptomer på sentral prematur seksuell utvikling (før 8-9 år): forekomst av akneutslag, tidlig utvikling av melkekjertler og tidlig menarche hos jenter; tidlig kjønnshår og stemmemutasjon hos gutter.
Når hamartomer dannes i den bakre delen av hypothalamus, kan det være unormaliteter i hjernens elektriske aktivitet, som i tidlig spedbarnsalder manifesterer seg ved anfall, og på et senere stadium (i alderen 4 til 7 år) ved epilepsi med fokale epileptiske anfall med plutselig latter eller ufrivillig gråt, atoniske og tonisk-kloniske anfall, samt anfall av aggresjon, hukommelse og kognitive problemer.
Et hypofysehamartom er et sporadisk forekommende godartet hypofyseadenom.
Middelaldrende voksne med Cowden syndrom kan ha en sjelden tumorlignende masse, et hamartom i lillehjernen, diagnostisert som dysplastisk cerebellært gangliocytom eller Lhermitte-Duclos sykdom. Symptomer kan være fraværende eller manifestere seg som hodepine, svimmelhet, nedsatt koordinasjon av bevegelser og lammelse av individuelle kranienerver.
Lymfeknutehamartom
Når cellene i glatt muskel og fettvev, samt blodårer og kollagent stroma i inguinale, retroperitoneale, submandibulære og cervikale lymfeknuter, vokser over, dannes et angiomyomatøst hamartom i en lymfeknute eller et nodulært angiomyomatøst hamartom – med delvis eller fullstendig erstatning av parenkymet. [ 25 ], [ 26 ]
Et hamartom i huden
I nærvær av tuberøs sklerose eller nevrofibromatose observeres forskjellige hamartomer i huden, oftest i form av hypopigmenterte flekker; kaffe- og melkeflekker; angiofibrom (på kinnene, haken, nasolabiale folder); shagreen-flekker av forskjellige lokaliseringer (som er bindevevsnevi); fibrøse plakk på pannen, hodebunnen eller nakken.
En sjelden dermatologisk manifestasjon av tuberøs sklerose (spesielt hos menn) er follikulocystisk og kollagen hamartom, karakterisert ved rikelig kollagenavsetning i dermis, konsentrisk perifollikulær fibrose og keratinfylte traktformede subkutane cyster sett ved histopatologisk undersøkelse. [ 27 ]
Når det gjelder hamartomer som består av melanocytter (celler som produserer pigmentet melanin), refererer de fleste eksperter også til forskjellige melanocytiske neoplasmer, spesielt medfødte melanocytiske nevi, som representerer en abnormalitet i embryogenesen.
Når det gjelder etiologi, er hamartomer bestående av vaskulært vev også hemangiomer i huden.
Pasienter med Peutz-Jeghers-Thuren syndrom har hamartom i form av flekkvis pigmentering av hud og slimhinner - lentiginosis periorificialis
Tilfeller av lineært papulært ektodermalt-mesodermalt hamartom (Hamartoma moniliformis) viser et lineært, kjøttfarget papulært utslett på hodet, nakken og øvre del av brystet.
Og et sebocytisk hamartom er et hamartom i talgkjertlene, les mer i publikasjonen - talgnevus.
Hamartom i øyet
Pigmenterte hamartomatøse lesjoner i iris ved nevrofibromatose type 1 og Watson syndrom – i form av nodulære klynger av dendrittiske melanocytter – defineres som irishamartomer eller Lisch-knuter. De er gjennomsiktige (vanligvis ikke synspåvirkende), avrundede kuppelformede gulbrune papler som stikker ut over irisoverflaten.
Og pasienter med juvenilt angiofibrom i nasofarynks og familiær adenomatøs polypose utvikler ofte kombinert hamartom i netthinnen og retinalt pigmentepitel – i form av en svart flekk på den sentrale (makula) delen av netthinnen. [ 28 ]
Et hamartom i nesen
Nasal hamartom defineres av spesialister som nasalt kondromesenkymalt hamartom eller nasalt kondrom, på grunn av godartet proliferasjon av respirasjonsepitel, submukosale kjertler og kondro-benmesenkym. De kliniske manifestasjonene avhenger av størrelsen og lokaliseringen av lesjonen og inkluderer: tett nese, vanskeligheter med nesepust og amming hos spedbarn, klar, vannaktig neseutflod og neseblødning. Et hamartom kan vokse med barnet og spre seg til øyehulene, noe som resulterer i forskyvning av øyeeplet fremover eller bakover, strabismus eller okulomotoriske forstyrrelser. [ 29 ]
Et hamartom hos et barn
Alle de ovennevnte hamartomatøse lesjonene i ulike organer og anatomiske strukturer er tilstede hos barn med tilsvarende syndromer.
Nyfødte har mesenkymalt hamartom i brystveggen eller bruskhamartom i ribbeina, som er faste, immobile masser som følge av fokal overvekst av normale skjelettelementer med brusk-, vaskulære og mesenkymale elementer. Dette hamartomet kan forårsake respirasjonssvikt og utvikling av respiratorisk distresssyndrom. Mesenkymalt hamartom i leveren er den nest hyppigste godartede leversvulsten hos barn. Denne tumorlignende formasjonen (oftere lokalisert i høyre organlapp) består av celler fra mesenkymalt stroma, hepatocytter og epitelceller i gallegangsslimhinnen. Det kliniske bildet inkluderer følbar masse i bukhulen, anoreksi og vekttap, og ved betydelig størrelse (opptil 10 cm og mer) dekker svulsten ekstrahepatiske galleganger og vena cava inferior, noe som fører til gulsott og ødem i underekstremiteter.
Et hamartom er et medfødt mesoblastisk nefrom (forekommer hos 1 av 200 000 spedbarn) som kan føre til oppblåsthet i magen hos den nyfødte med en følbar masse av tett konsistens i øvre høyre kvadrant av magen. Spedbarn kan også ha rask, overfladisk pust.
Sjeldne medfødte anomalier inkluderer fibrøst hamartom i spedbarnsalderen, som forekommer hos barn i de to første leveårene og presenterer seg som en smertefri nodulær masse i det subkutane vevet i armhulen, nakken, skulderen og underarmen, ryggen og brystet, låret, foten og de ytre kjønnsorganene.
Ekkrin angiomatøs hamartom hos et barn kan være tilstede ved fødselen eller manifestere seg i tidlig barndom. Denne godartede svulsten av hamartomatøs natur har vanligvis utseendet som blålige eller brunlige knuter og/eller plakk som skyldes proliferasjon av ekkrin svettekjertelvev og kapillærer i midtre og dype lag av dermis. Dette hamartomet kan forårsake lokalisert hyperhidrose og økt hårvekst.
Komplikasjoner og konsekvenser
Det er generelt enighet om at hamartomer sjelden kommer tilbake eller omdannes til ondartede svulster. De viser ofte få eller ingen symptomer, og noen ganger forsvinner de til og med over tid. Men i mer alvorlige tilfeller, og avhengig av dannelsesstedet, kan disse misdannelsene ha alvorlige komplikasjoner og konsekvenser.
Først og fremst kan et hamartom vokse til en slik størrelse at det begynner å presse på omkringliggende vev og organer, og forstyrre funksjonene deres.
Hjertehamartom hos barn kan føre til vedvarende hjerterytmeforstyrrelser, klaffefeil og nedsatt intrakardial blodstrøm med påfølgende hjertesvikt.
Komplikasjoner av hamartomatøse polypper i mage-tarmkanalen er gastrointestinal blødning, obstruksjon og intestinal invaginasjon (med mulig dødelig utgang). Og et stort nyrehamartom kan provosere nyreruptur.
Et hamartom i hjernen kan forårsake obstruktiv hydrocephalussyndrom.
Ved hypothalamiske og hypofysehamartomer kan produksjonen av somatotropisk hormon (veksthormon) være svekket, noe som kan føre til utvikling av hypofyseal nanisme (hypopituitarisme) hos barn. Hypothalamiske hamartomer hos barn kan også føre til medikamentresistent epilepsi.
Komplikasjoner av retinalt pigmentepitelhamartom er fulle av netthinne- og/eller synsnervedysfunksjon, makulaødem, neovaskularisering av årehinnen og netthinneavløsning.
Diagnostikk hamartomer
En viktig del av diagnostiseringen av hamartomer og relaterte syndromer er innsamling av anamnese, inkludert familiehistorie.
Laboratorietester inkluderer blodprøver: generelle kliniske; serumelektrolytter; lymfocyttprofil; kalsium-, kalium-, fosfat- og ureanivåer; og leverfunksjonstester. Om mulig utføres en finnålsaspirasjonsbiopsi av massen, siden histologisk undersøkelse er avgjørende for diagnose og valg av behandlingstaktikk.
Instrumentell diagnostikk gir visualisering av hamartomatøs tumorlignende formasjon og identifisering av dens nøyaktige lokalisering, som røntgen, angiografi, elektroencefalografi (EEG), ultralyd (sonografi), CT (computertomografi), PET (positronemisjonstomografi), MR (magnetisk resonansavbildning) brukes til.
Differensiell diagnose
Ved alle unormale masser er differensialdiagnose svært viktig. Dermed differensieres tuberkulom og hamartom; pulmonært hamartom og primær lungekreft, bronkogen karsinoid sykdom, metastatisk sykdom. Hjernehamartom bør skilles fra kraniofaryngeom og hypothalamus-kiasmatisk gliom. Og differensialdiagnosen av hamartom som medfødt mesoblastisk nefrom inkluderer Wilms-tumor (malignt nefroblastom), klarcellet sarkom i nyren og ossifiserende nyretumor hos spedbarn.
Hvem skal kontakte?
Behandling hamartomer
Hvis hamartomet er asymptomatisk og oppdages ved et uhell, er det ikke nødvendig med behandling, men det er nødvendig å overvåke dets "oppførsel" og pasientens tilstand. I andre tilfeller er terapien rettet mot å redusere symptomenes intensitet og forhindre komplikasjoner. For eksempel, ved hypothalamisk hamartom med symptomer på for tidlig pubertet, foreskrives visse medisiner som hemmer frigjøringen av visse hormoner. Hjertemedisiner brukes til å behandle symptomer på hjertesvikt hos pasienter med hjertehamartomer.
Kirurgisk fjerning av hamartomer er indisert for å bekrefte diagnosen og i tilfeller av medisinsk ukorrigerbare intense symptomer.
For eksempel kan lungehamartomer resekteres ved kilereseksjon og, i alvorlige tilfeller, ved fjerning av en lungelapp (lobektomi). Et brysthamartom kan også fjernes, og hvis det er stort, kan det være nødvendig med delvis eller fullstendig mastektomi.
Stereotaktisk radiofrekvenstermoablasjon eller laserablasjon kan brukes til å fjerne hamartomatøse polypper. Radiokirurgi med svært fokuserte gammastråler – gammakniv for hypothalamiske hamartomer eller astrocytiske hamartomer – brukes også.
Forebygging
Den eneste metoden for å forhindre utvikling av hamartomer kan betraktes som genetisk screening av barnets fremtidige foreldre.
Prognose
Den generelle prognosen for denne medfødte anomalien avhenger av lokaliseringen og størrelsen på neoplasmen, samt av komorbiditeter og pasientens generelle helsetilstand.