Medisinsk ekspert av artikkelen

Nye publikasjoner
Charcot-Marie-Tooths sykdom.
Sist anmeldt: 12.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
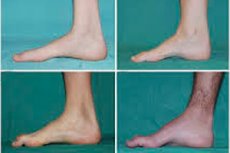
Peroneal muskelatrofi, Charcot-Marie-Tooth syndrom eller sykdom er en gruppe kroniske arvelige sykdommer med skade på perifere nerver.
I følge ICD-10, i avsnittet om sykdommer i nervesystemet, er koden for denne sykdommen G60.0 (arvelig motorisk og sensorisk nevropati). Den er også inkludert i listen over sjeldne sykdommer.
Epidemiologi
I følge klinisk statistikk er forekomsten av alle typer Charcot-Marie-Tooth sykdom per 100 000 av befolkningen 19 tilfeller (ifølge andre kilder, ett tilfelle per 2,5-10 000 av befolkningen).
CMT type 1 står for omtrent to tredjedeler av tilfellene (ett tilfelle per 5000 til 7000 innbyggere), og nesten 70 % av dem er assosiert med duplisering av PMP22-genet. Mer enn 1,2 millioner mennesker over hele verden lider av denne typen sykdom.
Forekomsten av CMT type 4 er anslått til 1–5 tilfeller per 10 000 barn. [ 1 ]
Fører til Charcot-Marie-Tooths sykdom
I henhold til klassifiseringen av polynevropatiske syndromer refererer peroneal (fibulær) muskelatrofi, Charcot-Marie-Tooth nevral amyotrofi eller Charcot-Marie-Tooth sykdom (forkortet CMT) til genetisk bestemte motorisk-sensoriske polynevropatier. [ 2 ]
Det vil si at årsakene til forekomsten er genetiske mutasjoner. Og avhengig av arten av genetiske avvik, skilles hovedtypene eller typene av dette syndromet: demyeliniserende og aksonale. Den første gruppen inkluderer Charcot-Marie-Tooth sykdom type 1 (CMT1), som oppstår på grunn av duplisering av PMP22-genet på kromosom 17, som koder for transmembran perifer myelinprotein 22. Som et resultat oppstår segmental demyelinisering av aksonskjeden (nervecelleprosesser) og en reduksjon i hastigheten på nervesignalledning. I tillegg kan mutasjoner være i noen andre gener.
Den aksonale formen er Charcot-Marie-Tooth sykdom type 2 (CMT2), som påvirker selve aksonene og er assosiert med patologiske endringer i MFN2-genet på lokus 1p36.22, som koder for membranproteinet mitofusin-2, som er nødvendig for mitokondriefusjon og dannelse av funksjonelle mitokondrielle nettverk inne i perifere nerveceller. Det finnes mer enn et dusin undertyper av CMT2 (med mutasjoner i spesifikke gener).
Det skal bemerkes at det for tiden er identifisert mer enn hundre gener hvis skade, overført ved arv, forårsaker ulike undertyper av Charcot-Marie-Tooth sykdom. For eksempel fører mutasjoner i RAB7-genet til CMT type 2B; endring av SH3TC2-genet (som koder for et av membranproteinene i Schwann-celler) forårsaker CMT type 4C, som manifesterer seg i barndommen og er preget av demyelinisering av motoriske og sensoriske nevroner (det finnes et dusin og et halvt former for type 4 av denne sykdommen).
En sjelden type 3 CMT (kalt Dejerine-Sottas syndrom) begynner å utvikle seg i tidlig barndom og er forårsaket av mutasjoner i PMP22, MPZ, EGR2 og andre gener.
Når CMT type 5 oppstår i alderen 5-12 år, observeres ikke bare motorisk nevropati (i form av spastisk paraparese i underekstremitetene), men også skade på syns- og hørselsnervene.
Muskelsvakhet og optisk atrofi (med synstap) samt balanseproblemer er typisk for CMT type 6. Og ved Charcot-Marie-Tooth sykdom type 7 er det ikke bare motorisk-sensorisk nevropati, men også en netthinnesykdom i form av retinitis pigmentosa.
X-bundet CMT eller Charcot-Marie-Tooth sykdom med tetraparese i lemmene (svakhet i bevegelse i både armer og ben), som er mer vanlig blant menn, er en demyeliniserende type og antas å skyldes en mutasjon i GJB1-genet på den lange armen av X-kromosomet, som koder for connexin 32, et transmembranprotein i Schwann-celler og oligodendrocytter som regulerer overføringen av nervesignaler. [ 3 ]
Risikofaktorer
Den viktigste risikofaktoren for CMT er en familiehistorie med sykdommen, det vil si hos nære slektninger.
Ifølge genetikere er risikoen for å få et barn som utvikler sykdommen 25 % dersom begge foreldrene er bærere av det autosomalt recessive genet for Charcot-Marie-Tooth sykdom. Og risikoen for at barnet er bærer av dette genet (men ikke har noen symptomer) er anslått til 50 %.
Ved X-bundet arv (når det muterte genet er på kvinnens X-kromosom), er det 50 % risiko for at moren gir genet videre til sønnen sin, som vil utvikle CMT. Sykdommen oppstår kanskje ikke når et jentebarn blir født, men datterens sønner (barnebarn) kan arve det defekte genet, og sykdommen vil utvikle seg.
Patogenesen
Ved enhver type Charcot-Marie-Tooth sykdom er patogenesen forårsaket av en arvelig anomali i perifere nerver: motoriske (bevegelse) og sensoriske (sensitive).
Hvis CMT-typen er demyeliniserende, fører ødeleggelsen eller defekten av myelinskjeden som beskytter aksonene i de perifere nervene til en nedgang i overføringen av nerveimpulser i det perifere nervesystemet - mellom hjernen, musklene og sanseorganene.
I den aksonale typen av sykdommen påvirkes selve aksonene, noe som negativt påvirker styrken til nervesignaler, noe som ikke er tilstrekkelig for full stimulering av muskler og sanseorganer.
Les også:
Hvordan overføres Charcot-Marie-Tooth syndrom? De defekte genene kan arves autosomalt dominant eller autosomalt recessivt.
Den vanligste typen, autosomal dominant arv, forekommer når det finnes én kopi av det muterte genet (båret av en av foreldrene). Og sannsynligheten for å overføre CMT til hvert født barn er anslått til 50 %. [ 4 ]
Ved autosomal recessiv arv trengs to kopier av det defekte genet (ett fra hver forelder som ikke viser noen tegn på sykdommen) for at sykdommen skal utvikle seg.
I 40–50 % av tilfellene forekommer autosomal dominant arvelig demyelinisering, dvs. CMT type 1; i 12–26 % av tilfellene aksonal CMT, dvs. type 2. Og i 10–15 % av tilfellene observeres X-bundet arv. [ 5 ]
Symptomer Charcot-Marie-Tooths sykdom
Vanligvis begynner de første tegnene på denne sykdommen å dukke opp i barndommen og ungdomsårene og utvikler seg gradvis gjennom livet, selv om syndromet kan gjøre seg kjent senere. Kombinasjonen av symptomer er variabel, og sykdomsprogresjonsraten, så vel som alvorlighetsgraden, er umulig å forutsi.
Typiske symptomer på den innledende fasen inkluderer økt generell tretthet; redusert tonus (svakhet) i musklene i føtter, ankler og leggben; mangel på reflekser. Dette gjør fotbevegelse vanskelig og fører til dysbasi (gangforstyrrelser) i form av høyere bena, ofte med hyppige snubling og fall. Tegn på Charcot-Marie-Tooth sykdom hos et lite barn kan inkludere uttalt klønethet og aldersuvanlige gangvansker forbundet med bilateral droppfot. Fotdeformiteter er også karakteristiske: høy fotbue (hulfot) eller alvorlig platføtter, buede (hammerformede) tær.
Ved gåing på tærne mot bakgrunn av muskelhypotoni, kan en nevrolog mistenke at barnet har type 4 CMT, der barn kanskje ikke kan gå i ungdomsårene.
Etter hvert som sykdommen utvikler seg, sprer muskelatrofi og svakhet seg til de øvre ekstremitetene, noe som gjør det vanskelig å utføre finmotorikk og normale håndoppgaver. Reduserte taktile sansninger og evnen til å føle varme og kulde, samt nummenhet i føtter og hender, indikerer skade på aksonene i sensoriske nerver.
Ved Charcot-Marie-Tooth sykdom type 3 og 6, som manifesterer seg i barndommen, observeres sensorisk ataksi (svekket koordinasjon av bevegelser og balanse), muskelrykninger og tremor, skade på ansiktsnerven, synsnerveatrofi med nystagmus og hørselstap.
I senere stadier kan det være ukontrollerbar risting (tremor) og hyppige muskelkramper; problemer med bevegelse kan føre til utvikling av smerter: muskel-, ledd-, nevropatisk.
Komplikasjoner og konsekvenser
Charcot-Marie-Tooth sykdom kan ha komplikasjoner og konsekvenser som:
- hyppigere forstuinger og brudd;
- kontrakturer forbundet med forkorting av periartikulære muskler og sener;
- skoliose (krumning av ryggraden);
- pusteproblemer – når nervefibrene som innerverer musklene i mellomgulvet er skadet:
- tap av evnen til å bevege seg selvstendig.
Diagnostikk Charcot-Marie-Tooths sykdom
Diagnosen omfatter klinisk undersøkelse, anamnese (inkludert familiehistorie), nevrologisk og systemisk undersøkelse.
Tester utføres for å sjekke bevegelsesomfang, følsomhet og senereflekser. Nerveledning kan vurderes ved hjelp av instrumentell diagnostikk – elektromyografi eller elektroneuromyografi. Ultralyd eller MR kan også være nødvendig. [ 6 ]
Genetisk testing eller DNA-testing for å oppdage de vanligste genetiske mutasjonene som forårsaker CMT i en blodprøve er begrenset fordi DNA-tester ikke er tilgjengelige for alle typer sykdommen for øyeblikket. For mer informasjon, se Genetisk testing.
I noen tilfeller utføres en biopsi av den perifere nerven (vanligvis den surale nerven).
Differensiell diagnose
Differensialdiagnose inkluderer andre perifere nevropatier, Duchennes muskeldystrofi, myelopatiske og myasteniske syndromer, diabetisk nevropati, myelopatier ved multippel og amyotrofisk lateral sklerose, Guillain-Barré syndrom, traume på peronealnerven og dens atrofi (inkludert ved klem mellom lumbalskivene i ryggraden), skade på lillehjernen eller thalamus, samt bivirkninger av cellegift (under behandling med cytostatika som vinkristin eller paklitaksel). [ 7 ]
Hvem skal kontakte?
Behandling Charcot-Marie-Tooths sykdom
I dag omfatter behandlingen av denne arvelige sykdommen treningsterapi (rettet mot å styrke og strekke muskler); ergoterapi (som hjelper pasienter med muskelsvakhet i hendene); og bruk av ortopediske hjelpemidler for å lette gange. Om nødvendig foreskrives smertestillende eller antikonvulsiva. [ 8 ]
Ved alvorlige platføtter kan osteotomi utføres, og ved hældeformasjon er kirurgisk korreksjon indisert – artrodese. [ 9 ]
Det pågår forskning på både den genetiske komponenten av sykdommen og behandlingsmetodene. Bruk av stamceller, visse hormoner, lecitin eller askorbinsyre har ennå ikke gitt positive resultater.
Men takket være nyere forskning kan det faktisk dukke opp noe nytt i behandlingen av Charcot-Marie-Tooths sykdom i nær fremtid. Dermed har det franske selskapet Pharnext siden 2014 utviklet, og siden midten av 2019 har kliniske studier pågått for legemidlet PXT3003 for behandling av CMT type 1 hos voksne, som undertrykker økt uttrykk av PMP22-genet, forbedrer myelinisering av perifere nerver og lindrer nevromuskulære symptomer.
Spesialister fra medisinselskapet Sarepta Therapeutics (USA) jobber med å lage en genterapi for Charcot-Marie-Tooth sykdom type 1. Denne terapien vil bruke et ufarlig adenoassosiert virus (AAV) av Dependovirus-slekten med et lineært enkelttrådet DNA-genom, som vil overføre NTF3-genet til kroppen, som koder for nevrotrofin-3 (NT-3)-proteinet som er nødvendig for funksjonen til Schwann-nerveceller.
Helixmith vil starte kliniske studier av den sørkoreanskutviklede genterapien Engensis (VM202) innen utgangen av 2020 for å behandle muskelsymptomer ved CMT type 1. [ 10 ]
Forebygging
Forebygging av CMT kan være genetisk veiledning av fremtidige foreldre, spesielt hvis noen i paret har en familiehistorie med sykdommen. Imidlertid har tilfeller av de novo punktgenmutasjoner blitt identifisert, det vil si i fravær av sykdommen i familiehistorien.
Under graviditet kan sannsynligheten for Charcot-Marie-Tooth sykdom hos det fremtidige barnet kontrolleres ved hjelp av chorionvillusbiopsi (fra 10. til 13. svangerskapsuke), samt en analyse av fostervann (ved 15-18 uker).
Prognose
Generelt sett avhenger prognosen for ulike typer Charcot-Marie-Tooth sykdom av den kliniske alvorlighetsgraden, men i alle tilfeller utvikler sykdommen seg sakte. Mange pasienter har funksjonshemminger, selv om dette ikke reduserer forventet levealder.