Medisinsk ekspert av artikkelen

Nye publikasjoner
Prioner - årsak til prionsykdommer
Last reviewed: 06.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
Langsomme virusinfeksjoner kjennetegnes av spesielle kriterier:
- en uvanlig lang inkubasjonsperiode (måneder, år);
- en spesifikk lesjon av organer og vev, først og fremst sentralnervesystemet;
- langsom, jevn progresjon av sykdommen;
- uunngåelig fatalt utfall.

Noen patogener som forårsaker akutte virusinfeksjoner kan også forårsake langsomme virusinfeksjoner. For eksempel forårsaker meslingevirus noen ganger SSPE, og røde hunder-virus forårsaker progressiv medfødt røde hunder og røde hunder-panencefalitt.
En typisk langsom virusinfeksjon hos dyr er forårsaket av visna/madi-viruset, som er et retrovirus. Det er årsaken til langsom virusinfeksjon og progressiv lungebetennelse hos sauer. Den hvite substansen i hjernen ødelegges, lammelse utvikler seg (visna - fortæring); kronisk betennelse i lungene og milten oppstår.
Sykdommer som ligner på langsomme virusinfeksjoner i sine trekk, er forårsaket av prioner – de som forårsaker prioninfeksjoner. Prionsykdommer er en gruppe progressive lidelser i sentralnervesystemet hos mennesker og dyr. Hos mennesker svekkes funksjonen til sentralnervesystemet, det forekommer personlighetsendringer og bevegelsesforstyrrelser. Symptomene på sykdommen varer vanligvis fra flere måneder til flere år, og ender med døden. Tidligere ble prioninfeksjoner ansett sammen med de såkalte som forårsaker langsomme virusinfeksjoner.
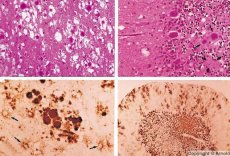
Noen agenser som forårsaker prionsykdommer akkumuleres først i lymfoidvev. Prioner, som kommer inn i hjernen, akkumuleres i store mengder, noe som forårsaker amyloidose (ekstracellulær dysproteinose, karakterisert ved avsetning av amyloid med utvikling av atrofi og sklerose i vevet) og astrocytose (proliferasjon av astrocytiske nevroglia, hyperproduksjon av gliafibre). Fibriller, aggregater av protein eller amyloid og svampformede forandringer i hjernen (overførbar svampform encefalopati) dannes. Som et resultat av dette endres atferd, koordinasjon av bevegelser svekkes, utmattelse med dødelig utgang utvikles. Immunitet dannes ikke. Prionsykdommer er konformasjonssykdommer som utvikler seg som et resultat av feil folding (brudd på korrekt konformasjon) av cellulært protein som er nødvendig for kroppens normale funksjon. Veiene for prionoverføring er varierte:
- fordøyelsesvei - infiserte produkter av animalsk opprinnelse, tilsetningsstoffer fra rå organer fra storfe, osv.:
- overføring gjennom blodoverføring, administrering av legemidler av animalsk opprinnelse, organ- og vevstransplantasjon, bruk av infiserte kirurgiske og tannlegeinstrumenter;
- overføring gjennom immunbiologiske preparater (infeksjon av 1500 sauer med PrP''' med hjerneformolvaksine fra syke sauer er kjent).
Patologiske prioner, som har kommet inn i tarmen, transporteres inn i blodet og lymfen. Etter perifer replikasjon i milten, blindtarmen, mandlene og annet lymfoidt vev, overføres de til hjernen gjennom perifere nerver (nevroinvasjon). Direkte penetrering av prioner inn i hjernen gjennom blod-hjerne-barrieren er mulig. Tidligere trodde man at sentralnervesystemet var det eneste vevet der patologiske prioner akkumuleres, men studier har dukket opp som har endret denne hypotesen. Det viste seg at akkumulering av prioner i milten er assosiert med økningen og funksjonen til follikulære dendrittiske celler.
 [
[ Egenskaper til prioner
Den normale cellulære isoformen av prionproteinet med en molekylvekt på 33–35 kDa bestemmes av prionproteingenet (priongenet PrNP er lokalisert på det 20. menneskelige kromosomet). Det normale genet forekommer på celleoverflaten (forankret i membranen av molekylets glykoprotein), og er følsomt for protease. Det regulerer overføringen av nerveimpulser, daglige sykluser, oksidasjonsprosesser, deltar i kobbermetabolismen i sentralnervesystemet og i reguleringen av stamcelledeling i benmargen. I tillegg finnes priongenet i milten, lymfeknuter, hud, mage-tarmkanal og follikulære dendrittiske celler.
Spredning av patologiske prioner
Transformasjonen av prioner til endrede former skjer når den kinetisk kontrollerte likevekten mellom dem forstyrres. Prosessen forsterkes av en økning i mengden patologisk (PrP) eller eksogent prion. PrP er et normalt protein forankret i cellemembranen. PrP' er et globulært hydrofobt protein som danner aggregater med seg selv og PrP'' på celleoverflaten: som et resultat transformeres PrP' til PrP'', og deretter fortsetter syklusen. Den patologiske formen av PrP''' akkumuleres i nevroner, noe som gir cellen et svampaktig utseende.
Kuru
Prionsykdom, tidligere vanlig blant papuanere (som betyr skjelving eller risting) i den østlige delen av øya Ny-Guinea. Sykdommens smittsomme egenskaper ble bevist av K. Gajdusek. Patogenet overføres via mat som følge av rituell kannibalisme - å spise den utilstrekkelig tilberedte, prioninfiserte hjernen til avdøde slektninger. Som følge av skade på sentralnervesystemet svekkes bevegelse og gange, frysninger og eufori ("latterdød") oppstår. Inkubasjonsperioden varer 5–30 år. Pasienten dør etter et år.
Creutzfeldt-Jakobs sykdom
Prionsykdom, som manifesterer seg som demens, syns- og lillehjerneforstyrrelser og bevegelsesforstyrrelser med dødelig utgang etter 4–5 måneders sykdom ved den klassiske varianten av Creutzfeldt-Jakobs sykdom og etter (3–14 måneder ved den nye varianten av Creutzfeldt-Jakobs sykdom. Inkubasjonstiden kan nå 20 år. Ulike smitteveier og årsaker til sykdommen er mulige:
- ved konsum av utilstrekkelig varmebehandlede animalske produkter, som kjøtt og hjerne fra kyr med bovin spongiform encefalopati;
- under vevstransplantasjon, som hornhinnetransplantasjon, blodtransfusjon, bruk av hormoner og andre biologisk aktive stoffer av animalsk opprinnelse, bruk av katgut, forurensede eller utilstrekkelig steriliserte kirurgiske instrumenter, prosektorale manipulasjoner;
- i tilfelle hyperproduksjon av PrR og andre tilstander som stimulerer prosessen med å omdanne PrR' til PrR".
Sykdommen kan også utvikles som følge av en mutasjon eller innsetting i prion-genregionen. Sykdommens familiære natur er vanlig på grunn av genetisk predisposisjon for Creutzfeldt-Jakobs sykdom. I den nye varianten av Creutzfeldt-Jakobs sykdom utvikles lidelsene i yngre alder (gjennomsnittsalder 28 år), i motsetning til den klassiske varianten (gjennomsnittsalder 65 år). I den nye varianten av Creutzfeldt-Jakobs sykdom akkumuleres unormalt prionprotein ikke bare i sentralnervesystemet, men også i lymforetikulært vev, inkludert mandlene.
Gerstmann-Sträussler-Scheinker syndrom
Arvelig prionsykdom, ledsaget av demens, hypotoni, svelgevansker (dysfagi), dysartri. Har ofte en familiær natur. Inkubasjonsperioden er fra 5 til 30 år. Sykdommen oppstår ved 50-60 års alder, og varigheten varierer fra 5 til 13 år.
Arvelig dødelig søvnløshet
En autoimmun sykdom med progressiv søvnløshet, sympatisk hyperreaktivitet (hypertensjon, hypertermi, hyperhidrose, takykardi), tremor, ataksi, multiklon, hallusinasjoner. Søvnen er alvorlig forstyrret. Døden inntreffer med progresjon av kardiovaskulær svikt.
Skrape
Skrapie (fra engelsk scrape - å skrape) er en prionsykdom hos sauer og geiter (skabb), som oppstår med skade på sentralnervesystemet, progressive bevegelsesforstyrrelser, alvorlig kløe i huden (skabb) og ender med dyrets død.
Bovin spongiform encefalopati
En sykdom hos storfe kjennetegnet av skade på sentralnervesystemet, svekket koordinasjon av bevegelser og uunngåelig død hos dyret. Sykdomsepidemien brøt først ut i Storbritannia. Den var forbundet med fôring av dyr med kjøtt- og beinmel som inneholdt patologiske prioner. Inkubasjonsperioden varierer fra 1,5 til 15 år. Hjernen, ryggmargen og øyeeplene hos dyr er mest infisert.
Laboratoriediagnostikk av prionsykdommer
Under diagnostikk observeres svampformede forandringer i hjernen, astrocytose (gliose) og fravær av inflammatoriske infiltrater. Hjernen farges for amyloid. Proteinmarkører for prionhjernesykdommer påvises i cerebrospinalvæsken (ved bruk av ELISA). Genetisk analyse av priongenet (PCR) utføres.
Forebygging av prionsykdommer
Autoklavering (ved 134 °C i 18 minutter; ved 121 °C i 1 time), forbrenning, ytterligere behandling med blekemiddel og en 1-N NaCl-løsning i 1 time anbefales for dekontaminering av instrumenter og miljøobjekter. For uspesifikk profylakse er det innført restriksjoner på bruk av legemidler av animalsk opprinnelse, og produksjon av hypofysehormoner av animalsk opprinnelse er forbudt. Transplantasjon av dura mater er begrenset. Gummihansker brukes ved arbeid med pasienters dialogvæsker.