Medisinsk ekspert av artikkelen

Nye publikasjoner
Cystisk fibrose
Sist anmeldt: 04.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
Cystisk fibrose er en genetisk autosomal recessiv monogen sykdom karakterisert ved en forstyrrelse i utskillelsen av eksokrine kjertler i vitale organer med skade primært på luftveiene og fordøyelsessystemet, alvorlig forløp og ugunstig prognose.
 [ 1 ]
[ 1 ]
Epidemiologi
Forekomsten av cystisk fibrose svinger mellom 1:2500 og 1:4600 nyfødte. Hvert år fødes det rundt 45 000 personer med cystisk fibrose over hele verden. Forekomsten av bærere av cystisk fibrose-genet er 3–4 %, med rundt 275 millioner mennesker over hele verden som bærere av dette genet, hvorav rundt 5 millioner bor i Russland og rundt 12,5 millioner i SNG-landene.
Fører til cystisk fibrose
Cystisk fibrose overføres autosomalt recessivt. Cystisk fibrose-genet er lokalisert i autosom 7, inneholder 27 eksoner og består av 250 000 nukleotidpar.
Et enkelt gen kan ha mange mutasjoner, som hver er spesifikk for en bestemt populasjon eller geografisk region. Mer enn 520 mutasjoner er beskrevet, hvorav den vanligste er delta-P-508, dvs. en substitusjon av aminosyren fenylalanin i posisjon 508.
Patogenesen
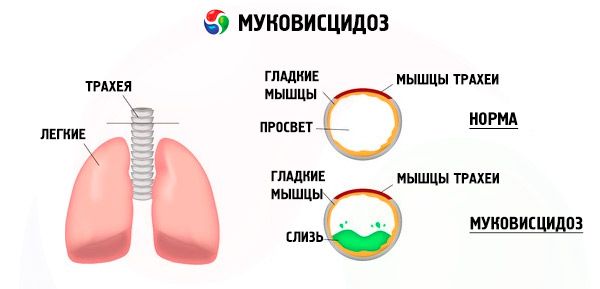
Mutasjoner i cystisk fibrose-genet forstyrrer strukturen og funksjonen til et protein kalt CFTR (cystisk fibrose transmembranregulator). Dette proteinet fungerer som en kloridkanal som er involvert i vann-elektrolyttutvekslingen av epitelceller i det bronkopulmonale systemet, mage-tarmkanalen, bukspyttkjertelen, leveren og reproduksjonssystemet. Som et resultat av forstyrrelsen av funksjonen og strukturen til CFTR-proteinet, akkumuleres kloridioner Cl⁻ inne i cellen. Dette fører til en endring i det elektriske potensialet i lumen i ekskretionskanalene, noe som letter strømmen av store mengder natriumioner (Na + ) fra kanalens lumen inn i cellen og ytterligere forbedret absorpsjon av vann fra det pericellulære rommet.
Som et resultat av disse endringene tykner utskillelsen av de fleste eksokrine kjertler, evakueringen forstyrres, noe som fører til uttalte sekundære lidelser i organer og systemer, mest uttalt i bronkopulmonale og fordøyelsessystemer.
En kronisk inflammatorisk prosess med varierende intensitet utvikler seg i bronkiene, funksjonen til det cilierte epitelet forstyrres kraftig, sputumet blir veldig tyktflytende, tykt, svært vanskelig å tømme ut, det stagnerer, bronkiolo- og bronkiektasi dannes, som over tid blir vanligere. Disse endringene fører til økt hypoksi og dannelse av kronisk pulmonal hjertesykdom.
Pasienter med cystisk fibrose er ekstremt disponert for utvikling av kronisk betennelse i det bronkopulmonale systemet. Dette skyldes uttalte forstyrrelser i det lokale bronkopulmonale forsvarssystemet (reduserte nivåer av IgA, interferon, fagocytisk funksjon av alveolære makrofager og leukocytter).
Alveolære makrofager spiller en viktig rolle i utviklingen av kronisk betennelse i det bronkopulmonale systemet. De produserer store mengder IL-8, noe som dramatisk øker nøytrofilkjemotaksi i bronkialtreet. Nøytrofiler akkumuleres i store mengder i bronkiene og utskiller, sammen med epitelceller, mange proinflammatoriske cytokiner, inkludert IL-1, 8, 6, tumornekrosefaktor og leukotriener.
En viktig rolle i patogenesen av skade på det bronkopulmonale systemet spilles også av den høye aktiviteten til enzymet elastase. Det skilles mellom eksogen og endogen elastase. Den første produseres av bakteriefloraen (spesielt Pseudomonas aeruginosa), den andre av nøytrofile leukocytter. Elastase ødelegger epitelet og andre strukturelle elementer i bronkiene, noe som bidrar til ytterligere forstyrrelse av mukociliær transport og rask dannelse av bronkiektasi.
Nøytrofile leukocytter utskiller også andre proteolytiske enzymer. Alfa-1-antipyrsin og en sekretorisk hemmer av leukoproteaser motvirker påvirkningen av proteolytiske enzymer og beskytter derfor det bronkopulmonale systemet mot deres skadelige innflytelse. Dessverre undertrykkes imidlertid disse beskyttende faktorene hos pasienter med cystisk fibrose av en betydelig mengde nøytrofil protease.
Alle disse omstendighetene bidrar til introduksjon av infeksjon i det bronkopulmonale systemet og utviklingen av kronisk purulent bronkitt. I tillegg bør det tas i betraktning at det defekte proteinet kodet av cystisk fibrose-genet endrer den funksjonelle tilstanden til bronkiepitelet, noe som favoriserer adhesjonen av bakterier til bronkiepitelet, primært Pseudomonas aeruginosa.
Sammen med patologien i det bronkopulmonale systemet forårsaker cystisk fibrose også alvorlig skade på bukspyttkjertelen, magen, tykktarmen og tynntarmen og leveren.
Symptomer cystisk fibrose
Cystisk fibrose manifesterer seg med ulike kliniske symptomer. Hos nyfødte kan sykdommen manifestere seg med mekoniumileus. På grunn av mangel på eller fullstendig fravær av trypsin blir mekonium svært tett, viskøst og akkumuleres i ileocekalregionen. Videre utvikles tarmobstruksjon, som manifesterer seg ved intens oppkast med galleblanding, oppblåst mage, manglende mekoniumutskillelse, utvikling av peritonittsymptomer og rask økning i kliniske manifestasjoner av alvorlig russyndrom. Barnet kan dø i løpet av de første levedagene hvis det ikke utføres øyeblikkelig kirurgisk inngrep.
I mindre alvorlige tilfeller er et karakteristisk tegn på cystisk fibrose rikelig, hyppig avføring, oljeaktig, med mye fett og en svært ubehagelig lukt. Hos 1/3 av pasientene observeres endetarmsprolaps.
Deretter fortsetter pasientene å oppleve tarmdysfunksjon, malabsorpsjonssyndrom, alvorlige fysiske utviklingsforstyrrelser og alvorlig hypovitaminose.
I løpet av det første eller andre leveåret oppstår symptomer på skade på bronkopulmonalt system (mild form av sykdommen), som manifesterer seg ved en hoste som kan være ekstremt uttalt og ligne kikhoste. Hosten er ledsaget av cyanose, kortpustethet og utskillelse av tykt sputum, først slimete og deretter purulent. Gradvis dannes et klinisk bilde av kronisk obstruktiv bronkitt og bronkiektasi, lungeemfysem og respirasjonssvikt. Barn er ekstremt utsatt for akutte respiratoriske virus- og bakterieinfeksjoner, noe som bidrar til forverring og progresjon av bronkopulmonal patologi. Utvikling av infeksjonsavhengig bronkial astma er mulig.
Hos skolebarn kan cystisk fibrose manifestere seg som "tarmkolikk". Pasienter klager over sterke paroksysmale magesmerter, oppblåsthet og gjentatt oppkast. Ved palpering av magen bestemmes tette formasjoner, lokalisert i tykktarmens projeksjon - avføring blandet med tykt, tett slim. Barn er svært utsatt for utvikling av hypokloremisk alkalose på grunn av overdreven utskillelse av salt med svette i varmt vær, mens "saltfrost" oppstår på barnets hud.
Sykdommer i bronkopulmonale system hos voksne
Skade på bronkopulmonale system hos pasienter med cystisk fibrose (lungeformen av sykdommen) er preget av utvikling av kronisk purulent obstruktiv bronkitt, bronkiektasi, kronisk lungebetennelse, lungeemfysem, respirasjonssvikt og pulmonal hjertesykdom. Noen pasienter utvikler pneumothorax og andre komplikasjoner av cystisk fibrose: atelektase, lungeabscesser, hemoptyse, lungeblødning og infeksjonsavhengig bronkial astma.
Pasienter klager over smertefull paroksysmal hoste med svært tyktflytende, vanskelig å separere mukopurulent sputum, noen ganger med en blanding av blod. I tillegg er kortpustethet ekstremt karakteristisk, først under fysisk anstrengelse, og deretter i hvile. Kortpustethet er forårsaket av bronkial obstruksjon. Mange pasienter klager over kronisk rhinitt forårsaket av polypose og bihulebetennelse. Betydelig svakhet, progressiv reduksjon i ytelse, hyppige akutte luftveisinfeksjoner er også karakteristiske. Ved undersøkelse rettes oppmerksomheten mot blek hud, poser i ansiktet, cyanose i synlige slimhinner og alvorlig kortpustethet. Med utviklingen av dekompensert pulmonal hjertesykdom oppstår ødem i bena. Fortykkelse av fingrenes terminale falanger i form av trommestikker og negler i form av urglass kan observeres. Brystet får en tønneformet form (på grunn av utviklingen av lungeemfysem).
Perkusjon av lungene avslører tegn på emfysem - en bokslyd, en skarp begrensning av mobiliteten i lungekanten og en senking av lungens nedre kant. Auskultasjon av lungene avslører hard pust med langvarig utånding, spredt tørr hvesing og fuktig middels og fin boblende hvesing. Ved alvorlig lungeemfysem svekkes pusten kraftig.
Ekstrapulmonale manifestasjoner av cystisk fibrose
Ekstrapulmonale manifestasjoner av cystisk fibrose kan være ganske uttalte og forekommer ofte.
Skade på bukspyttkjertelen
Insuffisiens av eksokrine funksjon i bukspyttkjertelen av varierende alvorlighetsgrad observeres hos 85 % av pasienter med cystisk fibrose. Ved mindre skade på bukspyttkjertelen er det fravær av dårlig fordøyelse og malabsorpsjonssyndromer, det er kun laboratoriemanifestasjoner av eksokrin insuffisiens (lave nivåer av trypsin og lipase i blodet og tolvfingertarmens innhold; ofte alvorlig steatoré). Det er kjent at for å forhindre dårlig fordøyelsessyndrom er det tilstrekkelig med utskillelse av bare 1 til 2 % av total lipase. Kun signifikante forstyrrelser i den eksokrine funksjonen manifesteres klinisk.
Under normale forhold produserer acini i bukspyttkjertelen en flytende sekresjon rik på enzymer. Når sekresjonen beveger seg langs ekskresjonskanalen, berikes den med vann og anioner, og den blir enda mer flytende. Ved cystisk fibrose, på grunn av en forstyrrelse i strukturen og funksjonen til transmembranregulatoren (kloridkanalen), mottar ikke bukspyttkjertelsekresjonen en tilstrekkelig mengde væske, den blir tyktflytende, og hastigheten på bevegelsen langs ekskresjonskanalen avtar kraftig. Sekresjonsproteinene avsettes på veggene i små ekskresjonskanaler, noe som resulterer i obstruksjon av dem. Etter hvert som sykdommen utvikler seg, utvikles det til slutt ødeleggelse og atrofi av acini - kronisk pankreatitt med eksokrin pankreatisk insuffisiens dannes. Dette gjenspeiles klinisk i utviklingen av dårlig fordøyelse og malabsorpsjonssyndromer. Bukspyttkjertelinsuffisiens er hovedårsaken til fettmalabsorpsjon ved cystisk fibrose, men dette observeres vanligvis med en betydelig lipasemangel. Forsher og Durie (1991) indikerer at ved fullstendig fravær av pankreatisk lipase brytes fett ned og absorberes med 50–60 %, noe som skyldes tilstedeværelsen av mage- og spyttlipaser (sublinguale), hvis aktivitet er nær den nedre grensen for normen. Sammen med forstyrrelsen av fettnedbrytning og -absorpsjon, skjer det en forstyrrelse av proteinnedbrytning og reabsorpsjon. Omtrent 50 % av proteinet som mottas med mat, går tapt med avføring. Karbohydratabsorpsjonen påvirkes i mindre grad til tross for mangel på α-amylase, men karbohydratmetabolismen kan forstyrres betydelig.
Skade på bukspyttkjertelen uttrykkes i utviklingen av dårlig fordøyelse og malabsorpsjonssyndrom med betydelig vekttap og rikelig med fet avføring.
Utviklingen av dårlig fordøyelse og malabsorpsjonssyndromer forenkles også av alvorlig dysfunksjon i tarmkjertlene, nedsatt utskillelse av tarmsaft og en reduksjon i innholdet av tarmenzymer i den.
Fordøyelsesproblemer og malabsorpsjonssyndromer kalles også den intestinale formen for cystisk fibrose.
Nedsatt endokrin funksjon i bukspyttkjertelen (diabetes mellitus) observeres hos pasienter med cystisk fibrose i de sene stadiene av sykdommen (hos 2 % av barn og 15 % av voksne)
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Lever- og galleveisskader
Hos 13 % av pasienter med blandede og intestinale former for cystisk fibrose utvikles levercirrhose. Det er mest typisk for mutasjonene W128X, delta-P508 og X1303K. Biliær levercirrhose med portalhypertensjon påvises hos 5–10 % av pasientene. I følge Welch, Smith (1995) påvises kliniske, morfologiske, laboratoriemessige og instrumentelle tegn på leverskade hos 86 % av pasienter med cystisk fibrose.
Mange pasienter med cystisk fibrose utvikler også kronisk kolecystitt, ofte kalklignende.
Dysfunksjon av kjønnskjertlene
Pasienter med cystisk fibrose kan oppleve azoospermi, som er årsaken til infertilitet. Redusert fertilitet er også typisk for kvinner.
Stages
Det er tre alvorlighetsgrader av pulmonal cystisk fibrose.
Den milde formen for cystisk fibrose er preget av sjeldne forverringer (ikke mer enn én gang i året); i perioder med remisjon er kliniske manifestasjoner praktisk talt fraværende, og pasientene er i stand til å jobbe.
Moderat alvorlighetsgrad - eksaserbasjoner observeres 2-3 ganger i året og varer i omtrent 2 måneder eller lenger. I eksaserbasjonsfasen er det en intens hoste med vanskelig å separere sputum, kortpustethet selv ved mindre fysisk anstrengelse, lav kroppstemperatur, generell svakhet, svette. Samtidig er det et brudd på bukspyttkjertelens eksokrine funksjon. I remisjonsfasen er ikke arbeidskapasiteten fullstendig gjenopprettet, kortpustethet under fysisk anstrengelse vedvarer.
Alvorlig forløp kjennetegnes av svært hyppige forverringer av sykdommen. Remisjoner er praktisk talt fraværende. I det kliniske bildet kommer alvorlig respirasjonssvikt, symptomer på kronisk pulmonal hjertesykdom, ofte dekompensert, hemoptyse er typisk, i forgrunnen. Betydelig vekttap observeres, pasientene er fullstendig ufør. Som regel er alvorlig bronkopulmonal patologi ledsaget av en kraftig uttrykt dysfunksjon i bukspyttkjertelen.
Skjemaer
- Bronkopulmonale lesjoner
- Gjentatt og tilbakevendende lungebetennelse med langvarig forløp.
- Abscessing av lungebetennelse, spesielt hos spedbarn.
- Kronisk lungebetennelse, spesielt bilateral.
- Bronkial astma som ikke er resistent mot tradisjonell behandling.
- Tilbakevendende bronkitt, bronkiolitt, spesielt ved Pseudomonas aeruginosa-kultur.
- Endringer i mage-tarmkanalen
- Meconium ileus og dens ekvivalenter.
- Syndrom med nedsatt intestinal absorpsjon av ukjent opprinnelse.
- Obstruktiv gulsott hos nyfødte med langvarig forløp.
- Levercirrose.
- Diabetes mellitus.
- Gastroøsofageal refluks.
- Kolelitiasis.
- Rektal prolaps.
- Endringer i andre organer og systemer
- Vekst- og utviklingsforstyrrelser.
- Forsinket seksuell utvikling.
- Mannlig infertilitet.
- Nesepolypper.
- Søsken fra familier med cystisk fibrose.
[ 24 ]
Komplikasjoner og konsekvenser
Komplikasjoner fra mage-tarmkanalen inkluderer:
- Diabetes mellitus utvikles hos 8–12 % av pasienter over 25 år.
- Fibroserende kolonopati.
- Mekoniumileus i nyfødtperioden (hos 12 % av nyfødte med cystisk fibrose), distalt intestinalt obstruksjonssyndrom, rektal prolaps, magesår og gastroøsofageal reflukssykdom.
Leverkomplikasjoner:
- Fettleversykdom (hos 30–60 % av pasientene),
- Fokal biliær cirrhose, multinodulær biliær cirrhose og tilhørende portal hypertensjon.
Portalhypertensjon fører noen ganger til død på grunn av spiserørsvarices.
Forekomsten av kolecystitt og gallestein er høyere hos pasienter med cystisk fibrose enn hos andre individer.
Forsinket pubertet og redusert fruktbarhet og andre komplikasjoner. De fleste menn har azoospermi og underutvikling av sædlederen.
Diagnostikk cystisk fibrose
Generell blodprøve - anemi av varierende alvorlighetsgrad er typisk, vanligvis normo- eller hypokrom. Anemi har en polyfaktoriell genese (redusert absorpsjon av jern og vitamin B12 i tarmen på grunn av utvikling av malabsorpsjonssyndrom). Leukopeni er mulig, med utvikling av purulent bronkitt og lungebetennelse - leukocytose, økt ESR.
Generell urinanalyse - ingen signifikante endringer, i sjeldne tilfeller observeres lett proteinuri.
Koprologisk undersøkelse - steatorrhea og kreatorrhea observeres. Becker (1987) anbefaler måling av kymotrypsin og fettsyrer i avføring. Før bestemmelse av kymotrypsin i avføring er det nødvendig å slutte å ta fordøyelsesenzymer minst 3 dager før undersøkelsen. Ved cystisk fibrose reduseres mengden kymotrypsin i avføring, og mengden fettsyrer økes (normal utskillelse av fettsyrer er mindre enn 20 mmol/dag). Det er nødvendig å ta hensyn til at økt utskillelse av fettsyrer med avføring også observeres ved:
- mangel på konjugerte fettsyrer i tynntarmen på grunn av leversvikt, obstruksjon av gallegangene, betydelig bakteriell kolonisering av tynntarmen (i dette tilfellet forekommer intensiv hydrolyse av gallesyrer);
- ileitt;
- cøliaki (med utvikling av malabsorpsjonssyndrom);
- enteritt;
- intestinale lymfomer;
- Whipples sykdom;
- matallergier;
- akselerert transitt av matmasser ved diaré av forskjellig opprinnelse, karsinoid syndrom, tyreotoksikose.
Biokjemisk blodprøve - reduserte nivåer av totalt protein og albumin, økte nivåer av alfa2- og gammaglobuliner, bilirubin og aminotransferaser (ved leverskade), redusert aktivitet av amylase, lipase, trypsin og jern- og kalsiumnivåer (ved utvikling av dårlig fordøyelsessyndrom, malabsorpsjon).
Sputumanalyse - tilstedeværelse av et stort antall nøytrofile leukocytter og mikroorganismer (under sputumbakterioskopi).
En studie av tynntarmens absorpsjonsfunksjon og bukspyttkjertelens eksokrine funksjon avslører betydelige forstyrrelser.
Røntgenundersøkelse av lungene - avslører endringer, hvis alvorlighetsgrad avhenger av alvorlighetsgraden og fasen av sykdommen. De mest karakteristiske endringene er:
- økt lungemønster på grunn av peribronkiale interstitielle forandringer;
- utvidelse av lungerøttene;
- bilde av lobulær, subsegmental eller til og med segmental atelektase i lungene;
- økt gjennomsiktighet i lungefeltene, hovedsakelig i de øvre delene, lav posisjon og utilstrekkelig mobilitet av diafragmaet, utvidelse av det retrosternale rommet (manifestasjon av lungeemfysem);
- segmental eller polysegmental infiltrasjon av lungevev (i utviklingen av lungebetennelse).
Bronkografi avslører endringer forårsaket av obstruksjon av bronkiene av viskøst sputum (fragmentering av bronkienes fylling med kontrastmiddel, ujevne konturer, fenomenet bronkial ruptur, en betydelig reduksjon i antall sidegrener), samt bronkoekgaser (sylindriske, blandede), lokalisert hovedsakelig i de nedre delene av lungene.
Bronkoskopi avslører diffus purulent bronkitt med rikelig tykt, viskøst sputum og fibrinøse filmer.
Spirometri - allerede i de tidlige stadiene av sykdommen avslører respirasjonssvikt av obstruktiv type (nedsatt FVC, FEV1, Tiffno-indeks), restriktiv (nedsatt FVC) eller, oftest, obstruktiv-restriktiv (nedsatt FVC, FVC, FEV1, Tiffno-indeks).
Gibson og Cooks svettetest (svetteelektrolytttest) innebærer stimulering av svette ved hjelp av pilokarpinelektroforese med påfølgende bestemmelse av klorider i svetten. Doerehuk (1987) beskriver testen slik. Pilokarpinelektroforese utføres på underarmen, den elektriske strømmen er 3 mA. Etter rensing av huden med destillert vann samles svetten opp ved hjelp av filterpapir plassert på det stimulerte området, dekket med gasbind for å forhindre fordampning fra det. Etter 30–60 minutter fjernes filterpapiret og elueres i destillert vann. Mengden oppsamlet svette måles. For å oppnå pålitelige resultater er det nødvendig å samle minst 50 mg (helst 100 mg) svette.
Hvis kloridkonsentrasjonen er over 60 mmol/l, anses diagnosen cystisk fibrose som sannsynlig; hvis kloridkonsentrasjonen er over 100 mmol/l, er den pålitelig; i dette tilfellet bør forskjellen i konsentrasjonen av klor og natrium ikke overstige 8–10 mmol/l. Hadson (1983) anbefaler at hvis natrium- og kloridinnholdet i svette er på grensen, bør en prednisolontest utføres (5 mg oralt i 2 dager, etterfulgt av bestemmelse av elektrolytter i svette). Hos personer som ikke lider av cystisk fibrose, synker natriumnivået i svette til den nedre normalgrensen; ved cystisk fibrose endres det ikke. En svettetest anbefales for alle barn med kronisk hoste.
Analyse av blodflekker eller DNA-prøver for større mutasjoner i cystisk fibrose-genet er den mest sensitive og spesifikke diagnostiske testen. Denne metoden er imidlertid bare egnet for land der delta-P508-mutasjonsraten er høyere enn 80 %. I tillegg er teknikken svært kostbar og teknisk kompleks.
Prenatal diagnose av cystisk fibrose utføres ved å bestemme isoenzymene av alkalisk fosfatase i fostervann. Denne metoden er mulig fra 18.–20. svangerskapsuke.
Hovedkriteriene for å diagnostisere cystisk fibrose er følgende:
- indikasjoner i anamnesen på forsinket fysisk utvikling i barndommen, tilbakevendende kroniske luftveissykdommer, dyspeptiske lidelser og diaré, tilstedeværelse av cystisk fibrose hos nære slektninger;
- kronisk obstruktiv bronkitt, ofte tilbakevendende, med utvikling av bronkiektasi og lungeemfysem, ofte tilbakevendende lungebetennelse;
- kronisk tilbakevendende pankreatitt med en markant reduksjon i eksokrin funksjon, malabsorpsjonssyndrom;
- økt klorinnhold i pasientens svette;
- infertilitet med bevart seksuell funksjon.
Vellykket diagnose og differensialdiagnose av cystisk fibrose forenkles ved å identifisere risikogrupper.
Screeningprogram for cystisk fibrose
- Generell analyse av blod, urin, sputum.
- Bakteriologisk analyse av sputum.
- Koprologisk analyse.
- Biokjemisk blodprøve: bestemmelse av totalt protein og proteinfraksjoner, glukose, bilirubin, aminotransferaser, alkalisk fosfatase, gamma-glutamyltranspeptidaser, kalium, kalsium, jern, lipase, amylase, trypsin.
- Studie av bukspyttkjertelens eksokrine funksjon og tarmens absorberende funksjon.
- Fluoroskopi og radiografi av lungene, CT-skanning av lungene.
- EKG.
- Ekkokardiografi.
- Bronkoskopi og bronkografi.
- Spirometri.
- Svettetest.
- Konsultasjon med en genetiker.
- Analyse av blodflekker eller DNA-prøver for større mutasjoner i cystisk fibrose-genet.
Hva trenger å undersøke?
Hvordan undersøke?
Hvilke tester er nødvendig?
Hvem skal kontakte?
Behandling cystisk fibrose
Typen og alvorlighetsgraden av symptomer på cystisk fibrose kan variere mye, så det finnes ingen typisk behandlingsplan; den er individualisert for hver enkelt person.
Terapien består av følgende terapeutiske tiltak:
- Pusteøvelser og postural drenasje bidrar til å bli kvitt tykt slim som samler seg i lungene. Noen teknikker for å rense luftveiene krever hjelp fra familiemedlemmer, venner eller en lungelege. Mange bruker en oppblåsbar brystvest som vibrerer med høy frekvens.

- Inhalasjonsmedisiner som har bronkodilaterende, drenerende (mukolytiske) og antibakterielle effekter (for eksempel fluorokinoloner).
- Preparater som inneholder pankreatiske enzymer for å forbedre fordøyelsen. Disse preparatene tas under måltider.
- Multivitaminer (inkludert fettløselige vitaminer).
I 2015 godkjente FDA et andre legemiddel for behandling av cystisk fibrose som er rettet mot et defekt protein kjent som CFTR. Det første legemidlet, en såkalt CFTR-modulator, ble godkjent i 2012. CFTR-modulatorer forventes å forlenge livet til noen mennesker med cystisk fibrose med flere tiår.
Kirurgi kan være nødvendig for å behandle følgende respiratoriske komplikasjoner:
- Pneumothorax, massiv tilbakevendende eller vedvarende hemoptyse, nesepolypper, vedvarende og kronisk bihulebetennelse.
- Mekoniumileus, tarminvaginasjon, endetarmsprolaps.
Lungetransplantasjon utføres i sykdommens terminale stadium.
Prognose
Gjennomsnittlig overlevelsesalder for pasienter med cystisk fibrose varierer fra 35 til 40 år. Gjennomsnittlig overlevelsesalder er høyere hos menn enn hos kvinner.
Med moderne behandlingsstrategier når 80 % av pasientene voksen alder. Cystisk fibrose begrenser imidlertid pasientens funksjonsevne betydelig. Det finnes fortsatt ingen kur for denne sykdommen.