Medisinsk ekspert av artikkelen

Nye publikasjoner
Martin-Bell syndrom
Sist anmeldt: 23.04.2024

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
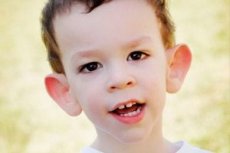
Martin-Bella syndromet ble beskrevet i 1943. Leger, hvis navn har blitt kalt. Sykdommen er en genetisk lidelse som består av mental retardasjon. I 1969 endringene som er karakteristiske for denne sykdommen i kromosom X (skjøthet i distal skulder) ble avslørt. I 1991 Forskere har oppdaget et gen som er ansvarlig for utviklingen av denne sykdommen. Denne sykdommen kalles også det "skjøre kromosom-X-syndromet." Sykdommen påvirker både gutter og jenter, men oftere (3 ganger) er gutter syk.
 [
[Epidemiologi
Martin-Bella syndromet er en ganske vanlig sykdom: det er 0,3-1,0 personer som lider av denne sykdommen per 1000 menn, og 0,2-0,6 per 1000 kvinner. Og barn med Martin-Bell syndrom er født på alle kontinenter med samme frekvens. Nasjonalt, hudfarge, snittet av øynene, levekårene, menneskets velvære påvirker åpenbart ikke sykdomsutbruddet. Hyppigheten av forekomsten er bare sammenlignbar med frekvensen av Downs syndrom (for 600-800 nyfødte, 1 sykdom). En femtedel av den mannlige bæreren av det forandrede genet er sunn, har ingen kliniske og genetiske abnormiteter, resten med tegn på mental retardasjon fra mild til alvorlig form. Blant kvinnelige bærere av pasienter litt mer enn en tredjedel.
Det skjøre X-kromosomsyndromet rammer omtrent 1 av 2500-4000 menn og 1 av 7000-8000 kvinner. Utbredelsen av bærere av sykdommen blant kvinners befolkning anslås å være 1 per 130-250 personer; utbredelsen av bærere blant menn er anslått til å være 1 per 250-800.
Fører til martin-Bell syndromet
Martin-Bella syndromet utvikler seg på grunn av fullstendig eller delvis opphør av produksjonen av et spesifikt protein av kroppen. Dette skyldes mangelen på respons fra et gen som FMR1, lokalisert i X-kromosomet. Mutasjon oppstår som et resultat av omorganisering av strukturen av genet fra ustabile strukturelle varianter av genstater (alleler), og ikke fra begynnelsen. Sykdommen overføres kun gjennom den mannlige linjen, som en mann kanskje ikke nødvendigvis er syk. Mannlige bærere, overføre genet til sine døtre i uendret form, så deres mentale retardasjon er ikke åpenbar. Med den videre overføringen av genet fra moren til sine barn, muterer genetet, og så vises alle tegnene som er karakteristiske for denne sykdommen.
Patogenesen
Er genmutasjoner i apparatet basert på patogenesen av syndromet Martin-Bell, noe som fører til blokkering av produksjonen av FMR-protein, protein, vital kroppen, spesielt i nerveceller og er til stede i forskjellige vev. Studier viser at FMR-proteiner er direkte involvert i reguleringen av oversettelser som forekommer i hjernevevet. Fraværet av dette proteinet eller dets begrensede produksjon av kroppen fører til mental retardasjon.
I patogenesen av sykdommen er nøkkelbrudd hypermetyleringen av genet, men det har ikke vært mulig å endelig bestemme mekanismen for utviklingen av denne lidelsen.
Sammen med dette ble stedets heterogenitet i patologien også avslørt, som er forbundet med polyallisme, så vel som polyl-fokusering. Tilstedeværelsen av alleliske varianter av utviklingen av sykdommen, som er forårsaket av eksistensen av punktmutasjoner, samt ødeleggelsen av FMRL-genet, bestemmes.
Også hos pasienter oppdages 2 følsomme for folsyrefraglete tripper i Z00 kb, og også 1,5-2 smp. Fra den skjøre tripleten, som inneholder genet FMR1. Mekanismen som forekommer i gener FRAXE, og FRAXF (de er identifisert i det ovenstående en skjør letter) mutasjoner korrelert med mekanismen til syndromlidelser Martin Bell. Denne mekanismen skyldes forplantning av GCC-, så vel som CGG-gjentakelser, under hvilke metylering av såkalte CpG-øyer oppstår. I tillegg til den klassiske form patologien eksisterer det også sjelden type 2, som skiller seg på grunn av utvidelse av trinukleotidet repetisjoner (i hann og hunn i meiose).
Det ble avdekket at i den klassiske form av syndromet har pasienten ikke et spesielt nukleocytoplasmisk protein av typen FMR1, som utfører funksjonen til å binde en rekke mRNAer. I tillegg fremmer dette proteinet dannelsen av et kompleks som bidrar til å utføre translasjonsprosesser inne i ribosomet.
Symptomer martin-Bell syndromet
Hvordan gjenkjenne sykdommen hos barn? Hva er de første tegnene? I de første månedene av barnets liv, kan symptomet på Martin-Bell ikke bli anerkjent, bortsett fra at noen ganger er det en nedgang i muskeltonen. Etter et år er klinikken til sykdommen mer åpenbar: barnet begynner å gå sent og snakke, noen ganger er talen helt fraværende. Han er hyperaktiv, uryddig svinger armene, redd for folkemengder og støy, sta, det er skarpe utbrudd av sinne, følelsesmessig ustabilitet, kramper oppstår, ikke går på øyekontakt. Pasienter med Martin-Bell syndrom sykdom produserer og utseende: ørene, utstående og stor, tung panne, avlangt ansikt, fremtredende hake, skjeling, brede hender og føtter. Endokrine lidelser er også karakteristiske for dem: ofte har tungvekt, fedme, menn store testikler, tidlig pubertet.

Blant pasientene med Martin-Bells syndrom er intelligensnivået svært forskjellig: fra en liten mental retardasjon til de alvorlige tilfellene. Hvis en normal person har en IQ på 100 i gjennomsnitt og et geni på 130, er 35-70 personer i fare for en sykdom.
Alle kliniske symptomer på patologi kan karakteriseres av en triade av grunnleggende manifestasjoner:
- oligofreni (IQ er 35-50);
- dysmorfofobi (fremspringende ører, samt prognatisme);
- makroorchidisme, som manifesterer seg etter utbruddet av puberteten.
Omtrent 80% av pasientene avslører også prolaps av bicuspid ventilen.
Men den fulde formen av syndromet manifesterer seg hos bare 60% av alle pasientene. I 10% er det bare mental retardasjon, mens i andre utvikler sykdommen med en annen kombinasjon av symptomer.
Blant de første tegn på sykdommen, manifestert allerede i tidlig alder:
- det syke barnet har betydelig mental retardasjon i forhold til utviklingen av andre jevnaldrende;
- oppmerksomhet og fokusforstyrrelser;
- sterk stædighet;
- barn begynner ganske sent å gå og snakke;
- det er hyperaktivitet og forstyrrelser i taleutvikling;
- veldig sterke og ukontrollerte bouts av sinne;
- kan utvikle en mutisme - dette er et komplett fravær av et barns tale;
- barnet føles sosial angst, er i stand til å få panikk på grunn av høy lyd eller andre sterke lyder;
- Barnet er ukontrollert og kaotisk vri på hendene;
- det er skinnhet, barnet er redd for å bo i steder av en stor mengde folk;
- fremveksten av ulike besettelser, en ustabil emosjonell tilstand;
- Barnet kan være motvillig til å etablere øynekontakt med mennesker.
Hos voksne observeres følgende symptomer på patologi:
- Spesielt utseende: Et langstrakt ansikt med tung panne, store fremspringende ører, en kraftig utstående hake;
- flate føtter, otitis og strabismus;
- puberteten oppstår ganske tidlig;
- fedme kan utvikle seg
- ganske ofte i Martin Bells syndrom er det mangler i hjerteutviklingen;
- hos menn er det en økning i testikler;
- leddene i leddene blir veldig mobile;
- Vekten, og også veksten øker kraftig.
Diagnostikk martin-Bell syndromet
For å diagnostisere Martin Bells syndrom må du kontakte en kvalifisert genetiker. Diagnosen er laget etter å ha utført spesifikke genetiske tester, som tillater å bestemme det defekte kromosomet.
Analyser
På et tidlig stadium av sykdomsutviklingen brukes en cytogenetisk metode hvor pasienten tar et stykke cellulært materiale, som folsyre blir tilsatt for å provosere endringer i kromosomene. Etter en viss tidsperiode avsløres regionen av kromosomet, hvor det vises en merkbar tynning - dette er tegn på tilstedeværelsen av det skjøre X-kromosomsyndromet.
Men denne analysen er ikke egnet for diagnose i sykdoms sentre, fordi nøyaktigheten er redusert på grunn av den store bruken av multivitaminer som inneholder folsyre.
Den integrerte diagnosen av Martin-Bells syndrom er en molekylærgenetisk undersøkelse som består i å bestemme antall såkalte trinucleotid-gjentagelser i genet.
Instrumental diagnostikk
En svært spesifikk metode for instrumentell diagnostikk er PCR (polymerasekjedereaksjon), som gjør det mulig å studere strukturen av aminosyrerester som finnes i X-kromosomet og dermed bestemme tilstedeværelsen av Martin Bell-syndromet.
Det finnes også en egen, enda mer spesifikk metode for diagnostisering av patologi - en kombinasjon av PCR og deteksjon ved hjelp av kapillærelektroforese. Denne metoden er svært nøyaktig og avslører en kromosomal patologi hos pasienter med primær form for ovarieinsuffisiens, samt ataksisk syndrom.
Bestem tilstedeværelsen av en defekt kan være etter diagnosen på EEG. Hos pasienter med denne sykdommen, observeres lignende bioelektrisk hjerneaktivitet.
Hvilke tester er nødvendig?
Differensiell diagnose
Differensierte metoder som bidrar til å mistenke et syndrom inkluderer:
- klinisk - 97,5% av pasientene har åpenbare tegn på mental retardasjon (moderat eller dypt); 62% hadde fremspringende store ører; 68,4% hadde en fremtredende fremtredende panne og panne; i 68,4% av guttene - testikler er forstørret, i 41,4% - talefunksjoner (talesatsen er ujevn, lydstyrken er ukontrollabel, etc.);
- cytogenetisk - blodet undersøkes for kulturen av lymfocytter, antall celler med brudd på X-kromosomet pr. 100 celler studert er bestemt;
- elektroencefalografi - endringene i hjernens elektriske impulser er spesifikke for Martin-Bell syndromet.
Hvem skal kontakte?
Behandling martin-Bell syndromet
Antidepressiva med psykostimulerende midler brukes til behandling av voksne pasienter. Prosessen med medisinering overvåkes kontinuerlig av en psykolog og en psykiater. I tillegg utfører private klinikker mikroinjeksjonsprosedyrer med rusmidler som Cerebrolysin (eller dets derivater), samt cytomediner (som for eksempel Solcoseryl eller Lidase).
Med utviklingen av ataksisk syndrom, brukes narkotika som fortynner blodet, så vel som nootropics. I tillegg er aminosyreblandinger og angioprotektorer foreskrevet. Kvinner med primær form for ovariesvikt foreskrives korrigerende behandling med fyto-rusmidler og østrogener.
Også, antagonister av glutaminreseptorer anvendes i behandling.
Tradisjonell for behandling av Martin-Bell syndrom er bruk av medisiner som påvirker symptomene på sykdommen, men ikke på grunn av det. Denne terapien består i utnevnelse av antidepressiva, neuroleptika, psykostimulerende midler. Ikke alle legemidler er indikert for bruk hos barn, så listen over medisiner er ganske begrenset. Neuroleptika som kan brukes etter 3 år (tidligste alder av avtalen) inkluderer haloperidol i dråper og tabletter, klorpromazin i oppløsning, pericyazin i dråper. Så er dosen av å ta haloperidol beregnet for barn, avhengig av kroppsvekten. For voksne administreres dosen individuelt. Godkjent innsiden, begynner med 0,5-5 mg 2-3 ganger daglig, deretter økes dosen gradvis til 10-15 mg. Når det er en forbedring, gå til en lavere dose for å opprettholde oppnådd tilstand. Med psykomotorisk stimulering utnevne 5-10 mg intramuskulært eller intravenøst, er det mulig flere repetisjoner på 30-40 minutter. Den daglige dosen bør ikke overstige 100 mg. Mulige bivirkninger i form av kvalme, oppkast, spasmer, økt trykk, arytmi etc. Spesielle forholdsregler bør følges av eldre mennesker. Tilfeller av plutselig hjertestans har blitt rapportert, og sen diskesi kan forekomme (forekomsten av ufrivillige bevegelser).
Antidepressiva øker aktiviteten til hjernestrukturer, lindrer deprimert stemning, spenning, øker stemningen. Disse legemidlene, anbefalt for inntak fra 5-8 år med Martin-Bell syndrom, inkluderer klomipromin, sertralin, fluoxegin, fluvoxamin. Så, fluoksetin tas under måltider innenfor 1-2 (fortrinnsvis om morgenen), begynner med 20 mg per dag, øker til 80 mg om nødvendig. Eldre mennesker anbefaler ikke en dose høyere enn 60 mg. Behandlingsforløpet bestemmes av legen, men ikke over 5 uker.
Mulige bivirkninger: Svimmelhet, angst, tinnitus, nedsatt appetitt, takykardi, hevelse, etc. Det skal tas forsiktighet ved ansettelse av eldre mennesker, med hjerte-og karsykdommer, diabetes mellitus.
Psykostimulerende midler - psykotrope stoffer, som brukes til å øke oppfatningen av ytre stimuli: forverre hørsel, respons, syn.
Som beroligende med nevoser, er angst, epileptiske anfall, anfall, diazepam foreskrevet. Det tas oralt, intravenøst, intramuskulært, rektalt (i endetarm). Det foreskrives individuelt, avhengig av alvorlighetsgraden av sykdommen, fra de minste doser på 5-10 mg, daglig - 5-20 mg. Behandlingens varighet er 2-3 måneder. For barn beregnes dosen med hensyn til kroppsvekt og individuelle egenskaper. Bivirkninger inkluderer sløvhet, apati, døsighet, kvalme, forstoppelse. Det er farlig å kombinere med alkohol, muligens vanedannende for stoffet.
Ved behandling av Martin-Bell syndrom tilfeller bedrer fast og innføring av produkter produsert på grunnlag av dyrematerialet (ledning): tserebrolizata, Cerebrolysin, tserebrolizat-M. Hovedkomponentene i disse legemidlene er peptider, som bidrar til proteinproduksjon i nevroner, og dermed påfyller det manglende protein. Cerebrolysin injiseres ved 5-10 ml, behandlingsforløpet består av 20-30 injeksjoner. Barn forskriver stoffet fra levetiden, injiseres intramuskulært hver dag for 1-2 ml i løpet av måneden. Mulige gjentatte økter med opptak. Bivirkninger i form av varme, kontraindisert hos gravide kvinner.
Det ble forsøkt å behandle sykdommen med folsyre, men bare det atferdsmessige aspektet ble forbedret (nivået av aggresjon, hyperaktivitet redusert, tale forbedret), men på det intellektuelle nivået var ingenting forandret. For å forbedre tilstanden til sykdommen foreskrevet folsyre, er metoder for fysioterapi, logopedisk, pedagogisk og sosial korreksjon angitt.
Litiumpreparater anses også som effektive, noe som bidrar til å forbedre pasientens tilpasning i det sosiale miljøet, samt kognitiv aktivitet. I tillegg regulerer de fortsatt sin oppførsel i samfunnet.
Bruk av urter i Martin-Bell syndrom er mulig som antidepressiva. Urter som bidrar til å avlaste spenning, angst, bedre søvn inkluderer valerian, peppermynt, timian, johannesurt, kamille. Infusjoner tilberedes som følger: 1 ts tørkede urter vil trenge et glass kokende vann, infusjoner insisterer minst 20 minutter, tatt hovedsakelig om natten før du går i seng eller om ettermiddagen. Et godt tillegg til dem vil være en skje med honning.
Fysioterapeutisk behandling
For å eliminere nevrologiske manifestasjoner, utføres spesielle fysioterapiprosedyrer - som bassengøvelser, muskelavslapping og akupunktur.
Operativ behandling
Et viktig behandlingsstadium betraktes også som plastikkirurgi - operasjoner som bidrar til å forbedre pasientens utseende. Plast av ekstremiteter og aurikler, og i tillegg til dette kjønnsorganene utføres. Det er også en korreksjon av gynekomasti med epispadia, og med det er det andre feil i utseende.
Forebygging
Den eneste metoden for å forebygge sykdommen er prenatal screening av gravide kvinner. Det er spesielle tester som tillater tidlig påvisning av forekomsten av patologi, hvorpå det anbefales å avslutte graviditeten. Som et alternativ er IVF brukt som kan hjelpe et barn til å arve et sunt kromosom X.
Profylaksen til pasienten avhenger av om mutasjonen av genet har oppstått igjen, eller ble arvet. For dette utføres molekylærgenetisk diagnose. I favør av "friskhet" mutasjoner er det faktum at slektninger av testen ikke avsløre "skjøre X-kromosom", og dermed risikoen for å få barn med syndromet Martin-Bell er svært liten. I familier hvor det er pasienter, vil testen bidra til å unngå gjentatte tilfeller.
Prognose
Prognosen for Martin-Bells syndrom for livet er gunstig, for gjenoppretting - nei. Forventet levetid avhenger av alvorlighetsgraden av sykdommen og tilhørende vices. Pasienten kan leve et liv med vanlig varighet. I alvorlige former for Martin-Bell syndrom truer livstruende funksjonshemming pasienter.
Levealder
Martin Bell-syndromet har ingen alvorlig negativ innvirkning på helse, slik at forventet levetid for de fleste som har funnet denne patologien, ikke er forskjellig fra standardindikatorene.