Medisinsk ekspert av artikkelen

Nye publikasjoner
Subakutt nekrotiserende encefalomyopati Leia
Sist anmeldt: 23.04.2024

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
 [
[Fører til av Leias syndrom
Sykdommen er basert på en enzymmangel, gi undervisning energi hovedsakelig av metabolske forstyrrelser pyrodruesyre og defekt elektrontransport i respirasjonskjeden. Pyruvat dehydrogenase-komplekset mangel utvikler (a-subenhet E1), pyruvat-carboxylase, kompleks 1 (NAD koenzym Q-reduktase), og komplekse 4 (cytokromoksydase) respirasjonskjeden.
Det ble funnet at defekter pyruvat, kompleks 1 (NAD koenzym Q-reduktase), og komplekse 4 (cytokromoksydase) respirasjonskjeden arves i en autosomal recessiv måte, defekter av pyruvat-dehydrogenase-komplekset (a-E1 subenhet) - X-bundet resessiv. Når mtDNA punkt mutasjoner som påvirker 6-ATPase-subenhet, mitokondriell arv karakteristikk. Oftest skjer mistsens mutasjon assosiert med utskifting av tymin til guanin eller cytosin i posisjon 8993 mtDNA. Mindre vanlig er en mutasjon på posisjon 9176 mtDNA. På grunn av det faktum at mutasjoner T8993G - grunnleggende mangel ved syndrom NARP, er beskrevet i familien med tilstedeværelsen av disse to sykdommer. Barn har også blitt beskrevet mtDNA mutasjon i posisjon 8344, som er funnet i syndromet MERRF.
Det foreslås at i tilfelle akkumulering av mutant mtDNA i de fleste mitokondrier utvikles en alvorlig løpet av Leia syndromet. I den mitokondrie-genetiske tilstanden til denne tilstanden, detekteres mutant mtDNA i 90% av alle mitokondrier. Pathogenese er forbundet med et brudd på energiproduksjon i celler og utvikling av melkesyreacidose.
Symptomer av Leias syndrom
De første tegnene på sykdommen debuterer i tidlig alder (1-3 år). Men det er tilfeller av sykdomsmanifestasjoner i to uker og på 6-7 år. Opprinnelig utviklet uspesifikke lidelser: psykomotorisk retardasjon, tap av matlyst, oppkast episoder, undervektige. I den påfølgende vekst nevrologiske symptomer: muskulær hypotoni eller dystoni med overgangen til hypertoni, beslag, myokloniske rykk eller tonisk-kloniske anfall, skjelving av lemmer, choreoathetosis, koordinasjonsforstyrrelser, redusert sene reflekser, letargi, tretthet. Serebral neurodegenerasjon har en progressiv natur. Plukke opp symptomer på pyramideformet og ekstrapyramidal sykdom, svekket svelge. Ofte er det slik endring av myndighet som ptose, oftalmoplegi, optisk atrofi, retinitis pigmentosa mindre. Noen ganger utvikler hypertrofisk kardiomyopati, er det episoder av takypné.
Sjelden fortsetter sykdommen i henhold til type akutt encefalopati. Mer karakteristisk er en kronisk eller subakutt strøm, noe som fører til dødelig utgang noen få år etter sykdomsutbruddet. Med en rask strømning (flere uker) skjer døden som følge av lammelse av luftveiene.
Diagnostikk av Leias syndrom
I en biokjemisk blodprøve oppdages laktatacidose på grunn av akkumulering av melkesyre og pyrodruesyrer i blodet og væsken, samt en økning i innholdet av alanin i blodet. Også nivået av ketonlegemer kan økes. I urinen oppdages økt utskillelse av organiske syrer: melk, fumarsyre etc. Karnitinnivåer i blod og vev reduseres ofte.
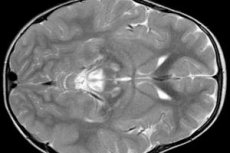
EEG-resultater viser fokale tegn på epileptisk aktivitet. Ifølge MR-data påvises en utvidelse av hjernens ventrikler, bilateral hjerneskade, kalsifisering av basalganglia (caudat kjerne, skall, svart stoff, blek ball). Det er også mulig å identifisere atrofi av hjernehalvene og hjernestoffene.
Morfologiske studie viser store forandringer i hjernen stoff: symmetrisk nekrose, demyelinering og svampaktig degenerasjon av hjernen, i første rekke midtdeler, bridge, basal ganglia, thalamus, synsnerven. Histologi innebærer cystisk degenerering av hjernevevet, astrocytic gliosis, neuronal død, øker antallet mitokondrier i cellene. I skjelettmuskel - akkumulering av lipid inneslutninger reduseres histokjemisk reaksjon på komplekser av 1, 4 respiratoriske kjede av mitokondrier subsarkolemmalnoe lunger, unormale mitokondrier med forstyrrelse av cristae. Fenomenet RRF er ofte ikke oppdaget.
Hvordan undersøke?
Hvilke tester er nødvendig?
Использованная литература