Medisinsk ekspert av artikkelen

Nye publikasjoner
Xeroderma pigmentosum
Sist anmeldt: 04.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
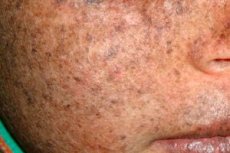
Xeroderma pigmentosum er en autosomal recessiv genetisk lidelse som påvirker DNA-reparasjon. Sykdommen er forårsaket av mutasjoner i gener som er involvert i reparasjon av DNA som er skadet av ultrafiolett stråling og andre giftige stoffer.
Oftest rammer denne sykdommen barn, som også kalles "nattens barn." Hyppige komplikasjoner av sykdommen er basalcellekarsinom og andre ondartede neoplasmer i huden, metastatisk malignt melanom og plateepitelkarsinom.
 [ 1 ]
[ 1 ]
Patogenesen
Mangel på UV-endonukleaseenzymer i pasientens celler (i fibroblaster) eller fullstendig fravær av disse spiller en viktig rolle i utviklingen av sykdommen. Dette enzymet er ansvarlig for reproduksjonen av DNA skadet av ultrafiolette stråler. Ifølge andre kilder spiller mangel på DNA-polymerase-1-enzymet en viktig rolle i utviklingen av sykdommen hos noen pasienter. Sykdommen utvikler seg oftest under påvirkning av stråler med en bølgelengde på 280-310 nm. Det antas at pigmentxerodermi utvikler seg på grunn av inntreden av forskjellige fotodynamiske stoffer i kroppen eller en økning i porfyriner i det menneskelige biologiske miljøet.
I de tidlige stadiene av sykdommen observeres hyperkeratose, atrofi av epidermisfokus, tynning av Malpighia-laget, en økning i melaningranuler i det basale cellelaget og kronisk betennelseinfiltrasjon i det øvre laget av dermis (hovedsakelig rundt blodårene). Deretter oppdages degenerative forandringer i kollagen og elastiske fibre, og i tumorstadiet avsløres tegn som er karakteristiske for hudkreft.
Symptomer xeroderma pigmentosum
Menn og kvinner er like rammet av denne sykdommen. Sykdommen begynner tidlig i barndommen om våren eller sommeren. Pasientens hud er spesielt følsom for sollys. Derfor oppstår det første utslettet i form av hyperpigmentering, lik en føflekk, på hudområder som er utsatt for sollys. Utslettet øker, erytem intensiveres, blir mørkebrunt, telangiektasi, angiomer og atrofi oppstår. Deretter vokser papillomer og vorter (hovedsakelig på huden i ansiktet og på halsen), flekker og sår oppstår. Som et resultat av arrdannelse etter sår blir nesen tynnere ("fuglenebb"), øyelokket vrenges ut. Papillomer utvikler seg vanligvis til ondartede svulster. Pigmentert xerodermi forekommer som regel sammen med spinocellulær kreft, melanosarkom.
Hva plager deg?
Stages
I det kliniske forløpet av xeroderma pigmentosum skilles det mellom fem stadier: inflammatorisk (erytematøs), hyperpigmentering, atrofi, hyperkeratose og ondartede svulster.
I løpet av den inflammatoriske (erytematøse) fasen oppstår hevelse, røde flekker og noen ganger blemmer og vesikler på hudområder som er utsatt for sollys (ansikt, nakke, øvre del av brystet, armer, hender). Det er nesten ikke noe utslett på områder som ikke er utsatt for sollys.
Ved hyperpigmentering oppstår det lysebrune, brune hyperpigmenterte flekker som ligner på føflekker i stedet for de røde flekkene.
Ved atrofi tørker huden ut, blir tynnere og rynker oppstår. Tallrike små, stellatformede telangiektasier og arr med en skinnende overflate observeres på huden på lepper og nese. På grunn av atrofi og arr utvikles mikrotomi (reduksjon av munnåpningen), ektropion, tynning av ører og nese, atresi av neseåpningen og munnen. Hos 80–85 % av pasientene er øynene påvirket: konjunktivitt, keratitt, skade på hornhinnen og slimhinnen, og svekket syn observeres. Dyskromi, telangiektasi, hyperkeratotiske forandringer og svulster observeres på huden på øyelokkene.
Ved hyperkeratose opptrer vortete svulster, papillom, keratoakantom, fibrom, angiofibriom og andre godartede svulster i de ovennevnte patologiske fokusene. Derfor inkluderer noen forskere pigmentxerodermi i gruppen av precancerøse sykdommer.
Etter 10–15 år fra sykdomsdebut oppstår ondartede hudsvulster (basaliom, endoteliom, angiosarkom) i atrofisk endrede foci og pigmentflekker. Svulstene ødelegges på kort tid, metastaserer til indre organer og fører til død. I indre organer og vev hos noen pasienter observeres generelle dystrofiske forandringer (syndaktelier i andre og tredje tå, tanndystrofi, fullstendig hårtap, etc.).
Skjemaer
Den nevrologiske formen av xeroderma pigmentosum manifesterer seg i to syndromer.
Reeds syndrom er karakterisert av kliniske manifestasjoner av xeroderma pigmentosum og mikrocefali, idiopati og langsom vekst av skjelettsystemet. I den nevrologiske formen av sykdommen er DNA-reparasjon vanskelig, og røntgenbehandling fører til en økning i de viktigste patologiske fokusene.
De Sinctis-X Cocchione syndrom er karakterisert av følgende kliniske tegn:
- utvikling av kliniske tegn på xeroderma pigmentosum på spesielt lysfølsom hud;
- tidlig forekomst av ondartede svulster;
- samtidig forløp av spastisk lammelse, mikrocefali og demens;
- medfødte misdannelser og dvergvekst;
- underutvikling av gonadene;
- hyppige spontanaborter;
- recessiv arveoverføring.
Ifølge noen hudleger er Santis-Cacchione syndrom ikke en uavhengig sykdom, men en alvorlig og fullt uttrykt klinisk manifestasjon av xeroderma pigmentosum.
Genetiske former
Typer |
Gen |
Lokus |
Beskrivelse |
Type A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) gruppe A – klassisk form |
Type B, II, XPB |
XPB |
2q21 |
XP Gruppe B |
Type C, III, XPC |
XPC |
3p25 |
XP Gruppe C |
Type D, IV, XPD |
XPD ERCC6 |
19q13.2–q13.3, 10q11 |
XP gruppe D eller De Sanctis-Cacchione syndrom |
Type E, V, XPE |
DDB2 |
23:00–12:00 |
XP Gruppe E |
Type F, VI, XPF |
ERCC4 |
16p13.3–p13.13 |
XP Gruppe F |
Type G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP gruppe G og COFS syndrom (cerebro-okulo-facioskjelettalt syndrom) type 3 |
Type V, XPV |
POLH |
6p21.1-p12 |
Variant Xeroderma Pigmentosa |
Hva trenger å undersøke?
Hvordan undersøke?
Hvem skal kontakte?
Behandling xeroderma pigmentosum
Antimalariamidler (delagyl, plaquenil, resoquin, etc.), som beskytter DNA mot ultrafiolette stråler, hemmer depolymeriseringsprosessen og reduserer hudens følsomhet (fotosensibilisering) for sollys. Disse legemidlene har betennelsesdempende og hyposensibiliserende egenskaper. Det anbefales å gjennomføre generell behandling sammen med vitaminbehandling (B1, B2, PP, B6, B12, A, E), antihistaminer (tavegil, difenhydramin, suprastin) og desensibiliserende midler (natriumtiosulfat, 10 % kalsiumklorid intravenøst 10 ml).
For lokal behandling brukes solkremer og salver.
Ved tumorformen av pigment-xerodermi brukes kirurgiske metoder, flytende nitrogen og laserstråler. For å beskytte deg mot sollys må du bruke løse klær, solhatt og hansker.
Prognose
De fleste pasientene (2/3) dør før de fyller 15 år. Noen pasienter kan leve til 40–50 år.
[ 20 ]