Medisinsk ekspert av artikkelen

Nye publikasjoner
Keratoderma: årsaker, symptomer, diagnose, behandling
Sist anmeldt: 07.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
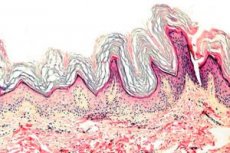
Keratodermi er en gruppe dermatoser som er preget av en forstyrrelse av keratiniseringsprosessen - overdreven horndannelse hovedsakelig på håndflatene og fotsålene.
Årsakene til og patogenesen til sykdommen er ikke fullt ut klarlagt. Forskning har slått fast at keratodermier er forårsaket av mutasjoner i genene som koder for keratin 6, 9, 16. Vitamin A-mangel, hormonelle dysfunksjoner, først og fremst i kjønnskjertlene, bakterielle og virusinfeksjoner er av stor betydning i patogenesen. De er et av symptomene på arvelige sykdommer og svulster i indre organer (parapsoriatiske keratodermier).
Symptomer. Det skilles mellom diffus (Unna-Tost keratodermi, Meleda keratodermi, Papillon-Lefèvre keratodermi, mutilerende keratodermi og syndromer som inkluderer diffus keratodermi som et av hovedsymptomene) og fokal (disseminert flekket keratodermi av Fischer-Buschke, akrokeratoelastoidose av Kosti, begrenset keratodermi av Bruhauer-Franzesthesti, lineær keratodermi av Fuchs, etc.) keratodermi.
Winy-Tost keratodermi (synonymer: medfødt iktyose i håndflater og fotsåler, Winy-Tost syndrom) overføres autosomalt dominant. Det er en diffus overdreven keratinisering av huden på håndflater og fotsåler (noen ganger bare sålene), som utvikler seg i løpet av de to første leveårene. Den patologiske hudens prosess begynner med en liten fortykkelse av huden på håndflater og fotsåler i form av en stripe av erytem med en lys farge på grensen til sunn hud. Over tid vises glatte, gulaktige hornlag på overflaten. Lesjonen sprer seg sjelden til håndleddsryggen eller fingrene. Hos noen pasienter kan det dannes overfladiske eller dype sprekker, og lokal hyperhidrose observeres. Hos pasienten som ble observert av forfatteren, led onkelen på mors side, broren og sønnen av Winy-Tost keratodermi.
Tilfeller av skade på negler (fortykkelse), tenner og hår ved Winy-Tost keratodermi i kombinasjon med ulike skjelettanomalier og patologier i indre organer, nervesystemet og det endokrine systemet er beskrevet.
Histopatologi. Histologisk undersøkelse avslører markert hyperkeratose, granulose, akantose og små inflammatoriske infiltrater i øvre del av dermis. Differensialdiagnose. Sykdommen må differensieres fra andre typer keratodermi.
Meleda keratodermi (synonymer: Meleda sykdom, medfødt progressivt akrokeratom, Siemens' palmoplantar transgradient keratose, Kogoys arvelige palmoplantar progressive keratose) arves autosomal recessivt. Denne formen for keratodermi er karakterisert av tykke, gulbrune hornlag med dype sprekker. En fiolett-lilla kant på flere millimeter er synlig langs kantene av lesjonen. Prosessen sprer seg vanligvis til baksiden av hender og føtter, underarmer og leggben. De fleste pasienter opplever lokal hyperhidrose. I denne forbindelse blir overflaten av håndflatene og fotsålene litt fuktig og dekket med svarte prikker (svettekjertelkanaler).
Sykdommen kan utvikle seg i alderen 15–20 år. Neglene tykner og deformeres.
Histopatologi. Histologisk undersøkelse avslører hyperkeratose, noen ganger akantose, og et kronisk inflammatorisk infiltrat i papillærdermis.
Differensialdiagnose. Melela keratodermi må skilles fra Unna-Tost keratodermi.
Keratoderma Papillon-Lefevre (synonym: palmoplantar hyperkeratose med periodontitt) arves autosomal recessivt.
Sykdommen manifesterer seg i 2.-3. leveår. Det kliniske bildet av sykdommen ligner på Melelas sykdom. I tillegg er det karakteristiske forandringer i tennene (unormaliteter i utbruddet av melketenner og permanente tenner med utvikling av karies, gingivitt, raskt progredierende periodontose med for tidlig tanntap).
Histopatologi. Histologisk undersøkelse avslører fortykkelse av alle lag i epidermis, spesielt hornlaget, og ubetydelige cellulære klynger av lymfocytter og histiocytter i dermis.
Differensialdiagnose. Sykdommen bør skilles fra andre keratodermier. Et viktig kjennetegn er den karakteristiske tannpatologien, som ikke finnes ved andre former for arvelig diffus keratodermi.
Keratoderma mutilans (synonymer: Fonwinkel syndrom, arvelig mutilerende keratom) er en type diffus keratodermi som arves autosomalt dominant. Den utvikler seg i løpet av det andre leveåret og er karakterisert av diffuse hornavleiringer på huden i håndflatene og fotsålene med hyperhidrose. Over tid dannes det snorlignende riller på fingrene, noe som fører til kontrakturer og spontan amputasjon av fingrene. Follikulær keratose uttrykkes på baksiden av hendene, samt i området rundt albue- og kneleddene. Negleplatene endres (ofte som urglass). Tilfeller av hypogonadisme, rubinrød alopecia, hørselstap og pachyonyki er beskrevet.
Histopatologi. Histologisk undersøkelse avslører alvorlig hyperkeratose, granulose, akantose og små inflammatoriske infiltrater i dermis, bestående av lymfocytter og histiocytter.
Differensialdiagnose. Når man skiller mutilerende keratodermi fra andre former for diffus keratodermi, bør man først og fremst ta hensyn til mutilasjonseffekten, som ikke er typisk for andre former. Ved differensialdiagnostikk av alle former for diffus keratodermi er det nødvendig å huske at det kan være et av hovedsymptomene på en rekke arvelige syndromer.
Behandling. Neotigazon er indisert i generell behandling av keratodermi. Dosen av legemidlet avhenger av alvorlighetsgraden av prosessen og er 0,3–1 mg/kg av pasientens vekt. I fravær av neotigazon anbefales vitamin A i en dose på 100 til 300 000 mg per dag over lengre tid. Ekstern behandling består av bruk av salver med aromatiske retinoider, keratolytiske og steroide midler.
 [
[ Hva plager deg?
Hva trenger å undersøke?
Hvordan undersøke?