Medisinsk ekspert av artikkelen

Nye publikasjoner
Polymyositis og dermatomyositis: årsaker, symptomer, diagnose, behandling
Sist anmeldt: 23.04.2024

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
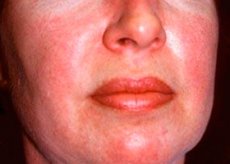
Polymyosit og dermatomyosit er sjeldne systemiske reumatiske sykdommer preget av inflammatoriske og degenerative muskelendringer (polymyositis) eller muskler og hud (dermatomyositis). Den mest spesifikke hud manifestasjonen er et heliotrope utslett.
Muskellesjonene er symmetriske og inkluderer svakhet, noe ømhet og etterfølgende atrofi av de proximale musklene i det øvre ekstremitetsbelte. Komplikasjoner kan omfatte indre organskader og malignitet. Diagnosen er basert på analysen av det kliniske bildet og evalueringen av muskelforstyrrelser ved å bestemme konsentrasjonene av de relevante enzymene, utføre MR, elektromyografi og biopsi av muskelvev. Behandlingen bruker glukokortikoider, noen ganger i kombinasjon med immunsuppressiva eller immunoglobuliner administrert intravenøst.
Kvinner er syke dobbelt så ofte som menn. Sykdommen kan oppstå i alle aldre, men oppdages hyppigere i intervallet fra 40 til 60 år; hos barn fra 5 til 15 år.
 [
[Hva forårsaker dermatomyosit og polymyositis?
Årsaken til sykdommen er ment å være en autoimmun reaksjon på muskelvev i genetisk predisponerte individer. Sykdommen er mer vanlig i nærvær av en belastet familiehistorie og bærere av noen HLA-antigener (DR3, DR52, DR56). Mulige oppstartsfaktorer er viral myosit og maligne neoplasmer. Det er rapporter om deteksjon i muskelceller av strukturer som ligner pikoravirus; I tillegg kan virus forårsake lignende sykdommer hos dyr. Maligniteter assosiasjon med dermatomyositt (mye mindre enn med polymyositt) tyder på at tumorvekst kan også være en utløsende mekanisme av sykdommer som følge av autoimmunitet løpe på felles tumorantigener og muskelvev.
I veggene i blodkarene i skjelettmuskulaturen detekteres forekomster av IgM, IgG og den tredje komplementkomponent; Dette gjelder spesielt hos barn med dermatomyositis. Pasienter med polymyositis kan også utvikle andre autoimmune prosesser.
Patofysiologi dermatomyositis og polymyositis
Patologiske endringer inkluderer celleskader og deres atrofi mot en bakgrunn av betennelse av varierende alvorlighetsgrad. Muskler i øvre og nedre ekstremiteter, samt ansikt, er mindre påvirket enn andre skjelettmuskler. Nederlaget for vinkelsytens muskulatur og øvre del av spiserøret, mindre ofte i hjertet, magen eller tarmen, kan føre til forstyrrelse i funksjonene til disse organene. Høye konsentrasjoner av myoglobin, forårsaket av rabdomyolyse, kan forårsake nyreskade. Det kan også være betennelsesforandringer i ledd og lunger, spesielt hos pasienter som har antisyntetiske antistoffer.
Symptomer på dermatomyosit og polymyosit
Utbruddet av polymyositis kan være akutt (særlig hos barn) eller subakutt (vanligvis hos voksne). Akutt viral infeksjon går noen ganger foran eller er startfaktoren for manifestasjonen av sykdommen, de hyppigste manifestasjoner som er svakhet i proksimale muskler eller hudutslett. Smerteopplevelser er mindre uttrykt enn svakhet. Kanskje utviklingen av polyarthralgi, fenomenet Raynaud, dysfagi, brudd på lungene, vanlige symptomer (feber, senking av masse, svakhet). Fenomenet Reynaud finnes ofte hos pasienter som har samtidig bindevevssykdommer.
Muskel svakhet kan utvikles i flere uker eller måneder. For den kliniske manifestasjonen av muskelsvikt må imidlertid minst 50% av muskelfibrene påvirkes (derfor viser tilstedeværelsen av muskel svakhet at myosit er blitt utviklet). Pasienter kan oppleve vanskeligheter med å løfte armene over skuldernivå, gå opp trappen, stiger opp fra sittende. På grunn av den utprøvde svakheten i bekkenmusklene og skulderbeltet, kan pasienter nitte på rullestol eller seng. Ved nederlag av nakkebøyle blir det umulig å rive av hodet fra puten. Nedfallet av muskler i strupehodet og øvre del av spiserøret fører til brudd på svelging og oppblåsthet. Muskler i nedre, øvre lemmer og ansikt påvirkes vanligvis ikke. Imidlertid er utviklingen av lemkontrakturer mulig.
Utslett er rapportert med dermatomyositt har vanligvis en mørk farge og erytematøst karakter. Periorbital hevelse av lilla farge (heliotrope utslett) er også karakteristisk. Hudutslett kan være litt hevet over huden og være glatt eller skjellete; lokalisasjon av lesjoner - pannen, nakke, skuldre, brystet, ryggen, underarmene, nedre partier av lårene bryn, kne-området, den mediale malleolus, den bakre flate av interfalangeale og metakarpofalangealleddene ledd, med den laterale side (Gottrona symptom). Mulig hyperemi av basen eller periferien av neglene. På huden av sideflatene av fingrene kan utvikle desquamative dermatitt, ledsaget av utseende av sprekker. Primære kutane lesjoner ofte løses uten konsekvenser, men kan føre til utvikling av sekundære forandringer i form av mørk pigmentering, atrofi, arrdannelse, eller vitiligo. Mulig dannelse av subkutane forkalkninger, spesielt hos barn.
Ca. 30% av pasientene utvikler polyarthralgi eller polyarthritis, ofte ledsaget av hevelse og felles effusjon. Likevel er alvorlighetsgraden av articular manifestasjoner lav. Oftere forekommer de når pasienter har antistoffer mot Jo-1 eller andre syntetaser.
Nederlaget for de indre organene (med unntak av svelget og øvre spiserøret) i polymyositis er mindre vanlig enn i andre reumatiske sykdommer (spesielt SLE og systemisk sklerodermi). Sjelden, spesielt med et antisyntetisk syndrom, manifesterer sykdommen som en interstitiell pneumonitt (i form av dyspné og hoste). Hjertearytmier og ledningsforstyrrelser kan utvikle seg, men de er vanligvis asymptomatiske. Manifestasjoner fra mage-tarmkanalen er vanligere hos barn som også lider av vaskulitt, og kan inkludere oppkast med en blanding av blod, melena og perforering av tarmen.
Hvor gjør det vondt?
Klassifisering av polymyositis
Det er 5 varianter av polymyositis.
- Primær idiopatisk polymyositis, som kan oppstå i alle aldre. Med det er det ingen hudlesjon.
- Primær idiopatisk dermatomyositis ligner den primære idiopatiske polymyositis, men med det er det en lesjon av huden.
- Polymyositis og dermatomyositis assosiert med ondartede neoplasmer kan forekomme hos pasienter i alle aldre; oftest observert hos eldre pasienter, så vel som hos pasienter med andre bindevevssykdommer. Utviklingen av ondartede neoplasmer kan observeres både innen 2 år før og innen 2 år etter debut av myosit.
- Barns polymyositis eller dermatomyositis er assosiert med systemisk vaskulitt.
- Polymyositis og dermatomyositis kan også forekomme hos pasienter som lider av andre bindevevssykdommer, oftest med progressiv systemisk sklerose, blandet bindevevssykdom og SLE.
Inkludering av muskel i stammen i gruppen av polymyositis av myositis er feil, siden sistnevnte er en egen sykdom preget av kliniske manifestasjoner som ligner på kronisk idiopatisk polymyositis. Imidlertid utvikler det i den eldre, ofte påvirker de distale muskler i elementseksjoner (for eksempel øvre og nedre lemmer), har en lengre varighet mindre reagerer på behandling, og kjennetegnes ved typisk histologisk mønster.
Diagnose av dermatomyosit og polymyositis
Polymyositt bør mistenkes dersom pasienten klager om den svakhet av de proksimale muskler, ledsaget av deres ømhet eller uten. Testing for dermatomyositt er nødvendig for pasientene klaget over et utslett som ligner heliotrop, eller symptom Gottrona, så vel som i pasienter med manifestasjoner av polymyositt, kombinert med noen hudlesjoner, dermatomyositt hensiktsmessig. De kliniske manifestasjonene av polymyositis og dermatomyositis kan lignes på systemisk sklerose eller, sjeldnere, SLE eller vaskulitt. Diagnosens pålitelighet økes ved å matche det største antallet av disse fem kriteriene:
- svakhet av proksimale muskler;
- karakteristiske hudutslett;
- økt aktivitet av muskelvevsenzymer (kreatinkinase eller, i fravær av økning i aktivitet, aminotransferaser eller aldolase);
- karakteristiske endringer i myografi eller MR;
- karakteristiske histologiske endringer i biopsi av muskelvev (absolutt kriterium).
En muskelbiopsi kan utelukke noen klinisk lignende forhold, som muskel torso muskel og rhabdomyolyse forårsaket av en virusinfeksjon. Endringene som ble avslørt ved histologisk undersøkelse, kan være forskjellige, men typisk er kronisk betennelse, fokal degenerasjon og regenerering av muskler. Før utbruddet av potensielt giftig behandling, bør det foretas en nøyaktig diagnose (vanligvis ved histologisk verifisering). Med MR er det mulig å identifisere fokus på ødemer og betennelser i musklene etterfulgt av målrettet biopsi.
Laboratoriestudier tillate å forsterke eller omvendt, for å eliminere mistanke om tilstedeværelse av sykdommen, og er også nyttig for å vurdere dens alvorlighetsgrad, mulig kombinasjon med andre diagnostikk og patologi lignende komplikasjoner. Til tross for at antinukleære antistoffer oppdages hos noen pasienter, er dette fenomenet mer typisk for andre bindevevssykdommer. Omtrent 60% av pasientene har antistoffer mot nukleært antigen (PM-1) eller hele celler av thymus og Jo-1. Rollen av autoantistoff i patogenesen av sykdommen er uklar, selv om det er kjent at antistoffer mot Jo-en er spesifikk markør antisintetaznogo syndrom bestående fibrosing alveolitt, lungefibrose, artritt, Raynauds fenomen.
Periodisk vurdering av kreatinkinaseaktivitet er nyttig for overvåking av behandling. Likevel, med alvorlig muskelatrofi, kan enzymets aktivitet være normal, til tross for tilstedeværelsen av kronisk aktiv myosit. MR-data, muskelbiopsier eller høye verdier av kreatinkinasaktivitet bidrar ofte til å differensiere tilbakefall av polymyosit og myopati indusert av glukokortikoider.
Fordi mange pasienter har udiagnostisert kreft, anbefaler noen forfattere screening alle voksne med dermatomyositt, og personer som lider av polymyositt, i en alder av 60 som følger: fysisk undersøkelse, vkpyuchayuschy bryst eksamen, bekken eksamen og undersøkelse av endetarmen ( inkludert studier av avføring for latent blod); klinisk blodprøve; biokjemiske blodprøver; mammografi; bestemmelse av kreftembryonisk antigen; generell analyse av urin; bryst røntgen. Behovet for slike screening pasienter yngre alder, har ingen kliniske symptomer på kreft, noen forfattere tviler.
Hva trenger å undersøke?
Hvordan undersøke?
Behandling av dermatomyosit og polymyosit
Før du stopper betennelse, er det nødvendig å begrense fysisk aktivitet. Glukokortikoider er førstelinje medisiner. I det akutte stadiet av sykdommen trenger voksne pasienter prednisolon (inne) i en dose på 40 til 60 mg per dag. Regelmessig gjenkjenning av kreatinkinaseaktivitet er en tidlig indikator for effekt: i de fleste pasienter forekommer reduksjon eller normalisering innen 6 til 12 uker etter økningen av muskelstyrken. Etter normalisering av enzymaktiviteten, reduseres dosen av prednisolon: først ca. 2,5 mg per dag i løpet av uken, deretter raskere; Når aktiviteten til muskel enzymer øker, øker dosen av hormonet igjen. Gjenopprettede pasienter kan uten glukokortikoider, men oftere krever voksne pasienter med langvarig glukokortikoidbehandling (10-15 mg prednisolon per dag). Den første dosen prednisolon for barn er 30-60 mg / m 2 en gang daglig. Hvis det er remisjon> 1 år, kan barn ha opphørt glukokortikoidbehandling.
I noen tilfeller forekommer det plutselig økning i muskelsvikt hos pasienter som får høye doser glukokortikoider, noe som kan skyldes utviklingen av glukokortikoid-myopati.
Når utilstrekkelig respons på glukokortikoid behandling og også i utviklingen av glukokortikoid myopati eller andre komplikasjoner som krever reduksjon av dosen eller uttak prednisolon, bruker immunundertrykkende midler (metotreksat, cyklofosfamid, azatioprin, cyklosporin). Noen pasienter kan bare motta metotreksat (vanligvis i høyere doser enn ved behandling av RA) i mer enn 5 år. Intravenøse immunoglobuliner kan være effektiv i pasienter som ikke responderer på medikamentterapi, men deres anvendelse øker kostnadene for behandlingen.
Myositis assosiert med primære og metastaserende tumorer, samt myosit av stammen, er vanligvis mer ildfast mot glukokortikoidbehandling. Utviklingen av remisjon forbundet med ondartede svulster av myosit er mulig etter fjerning av svulsten.
Hvilken prognose har dermatomyosit og polymyositis?
Langvarig remisjon (og jevn klinisk gjenoppretting) i 5 år er registrert hos mer enn halvparten av behandlede pasienter; hos barn er denne indikatoren høyere. Tilbakeblikk kan imidlertid utvikles når som helst. Overordnet femårsoverlevelse er 75%, høyere hos barn. Årsakene til død hos voksne er alvorlig og progressiv muskelsvikt, dysfagi, nedsatt ernæring, aspirasjons lungebetennelse eller respirasjonsfeil på grunn av lungeinfeksjoner. Poliomyositis er mer alvorlig og resistent mot behandling i nærvær av skade på hjerte og lunger. Død hos barn kan oppstå på grunn av tarmvaskulitt. Den totale prognosen for sykdommen bestemmes også av tilstedeværelsen av ondartede neoplasmer.