Medisinsk ekspert av artikkelen

Nye publikasjoner
T-celle-lymfomer i huden
Last reviewed: 04.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
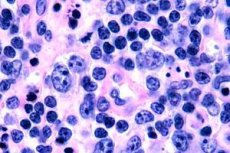
T-cellelymfomer er oftest registrert hos eldre, selv om isolerte tilfeller av sykdommen er observert selv hos barn. Menn er syke dobbelt så ofte som kvinner. T-cellelymfomer er epidermotrope av natur.
Fører til T-celle-lymfomer i huden
Årsakene til og patogenesen til kutane T-cellelymfomer er ikke fullt ut forstått. For tiden anser de fleste forskere humant T-celleleukemivirus type 1 (HTLV-1) I som den viktigste etiologiske faktoren som initierer utviklingen av ondartede T-cellelymfomer i huden. Sammen med dette diskuteres rollen til andre virus i utviklingen av T-cellelymfom: Epstein-Barr-virus, herpes simplex type 6. Hos pasienter med T-cellelymfom finnes virus i huden, perifert blod og Langerhans-celler. Antistoffer mot HTLV-I påvises hos mange pasienter med mycosis fungoides.
En viktig plass i patogenesen av T-cellelymfomer spilles av immunopatologiske prosesser i huden, hvorav den viktigste er den ukontrollerte proliferasjonen av klonale lymfocytter.
Cytokiner produsert av lymfocytter, epitelceller og celler i makrofagsystemet har proinflammatoriske og proliferative effekter (IL-1, ansvarlig for lymfocyttdifferensiering; IL-2 - T-cellevekstfaktor; IL-4 og IL-5, som øker tilstrømningen av eosinofiler inn i lesjonen og deres aktivering, etc.). Som et resultat av tilstrømningen av T-lymfocytter inn i lesjonen dannes Pautrier-mikroabscesser. Samtidig med økningen i lymfocyttproliferasjon undertrykkes aktiviteten til antitumorforsvarsceller: naturlige drepere, lymfocytotoksiske lymfocytter, dendrittiske celler, spesielt Langerhans-celler, samt cytokiner (IL-7, IL-15, etc.) - tumorvekshemmere. Rollen til arvelige faktorer kan ikke utelukkes. Tilstedeværelsen av familiære tilfeller og hyppig påvisning av noen histokompatibilitetsantigener (HLA B-5 og HLA B-35 - ved svært ondartede hudlymfomer, HLA A-10 - ved mindre aggressive lymfomer, HLA B-8 - i den erytrodermiske formen av mycosis fungoides) bekrefter dermatosens arvelige natur.
Kliniske observasjoner indikerer en mulig transformasjon av langvarige kroniske dermatoser (nevrodermatitt, atopisk dermatitt, psoriasis, etc.) til mycosis fungoides. Nøkkelfaktoren er den langvarige persistensen av lymfocytter i betennelsesfokuset, noe som forstyrrer immunovervåkingen og fremmer fremveksten av en klon av ondartede lymfocytter og dermed utviklingen av en ondartet proliferativ prosess.
Påvirkningen av fysiske faktorer på kroppen, som solinnstråling, ioniserende stråling og kjemiske stoffer, kan føre til fremveksten av en klon av «genotraumatiske» lymfocytter som har en mutagen effekt på lymfoide celler og utvikling av malignitet i lymfocytter.
Derfor kan T-cellelymfomer betraktes som en multifaktoriell sykdom som begynner med aktivering av lymfocytter under påvirkning av ulike kreftfremkallende, "genotraumatiserende" faktorer og fremveksten av en dominant T-celleklon. Alvorlighetsgraden av immunovervåkingsforstyrrelsen, klonen av ondartede lymfocytter, bestemmer de kliniske manifestasjonene (flekkete, plakk- eller tumorelementer) av T-cellelymfomer.
Patogenesen
I det tidlige stadiet av mycosis fungoides observeres akantose med brede prosesser, hyperplasi og kompaktering av basale keratinocytter, vakuolær degenerasjon av noen basalceller, atypiske mitoser i forskjellige lag av epidermis, epidermotropisme av infiltratet med penetrering av lymfocytter inn i epidermis. I dermis observeres små infiltrater rundt karene, bestående av enkle mononukleære celler med hyperkrome kjerner - "mykotiske" celler. I det andre stadiet observeres en økning i alvorlighetsgraden av dermal infiltrat og epidermotropisme av infiltratcellene, som et resultat av at ondartede lymfocytter trenger inn i epidermis og danner klynger i form av Potriers mikroabscesser. I det tredje, tumorstadiet, observeres massiv akantose og mindre atrofi av epidermis, samt økt infiltrasjon av epidermis av tumorlymfocytter, som danner flere Potriers mikroabscesser. Det massive infiltratet er lokalisert gjennom hele tykkelsen av dermis og dekker en del av hypodermis. Blastformer av lymfocytter observeres.
Kutant stort anaplastisk T-celle lymfom
Det er representert av en gruppe lymfoproliferative prosesser karakterisert ved tilstedeværelsen av proliferater fra atypiske klonale store anaplastiske CD30+ T-celler. Som regel utvikler det seg sekundært i tumorstadiet av mycosis fungoides eller ved Sezary syndrom, men kan utvikle seg uavhengig eller med spredning av systemiske lymfomer av denne typen. Klinisk tilsvarer slike lymfomer den såkalte dekapiterte formen av mycosis fungoides i form av enkle eller flere noder, vanligvis gruppert.
Histologisk opptar proliferatet nesten hele dermis med eller uten epidermotropisme i tilfelle epidermal atrofi.
Cytologisk kan tumorceller variere i størrelse og form. Basert på disse egenskapene skilles det mellom mellom- og storcellet pleomorf T-cellelymfom med kjerner av forskjellige uregelmessige konfigurasjoner - konvolverte, flerflikete, med tett kromatin, en veldefinert nukleol og ganske rikelig cytoplasma; immunoblastisk - med store runde eller ovale kjerner med tydelig karyoplasma og én sentralt plassert nukleol; anaplastisk - med stygge, veldig store celler med kjerner av uregelmessig konfigurasjon og rikelig cytoplasma. Fenotypisk tilhører hele denne gruppen T-hjelperlymfomer og kan være CD30+ eller CD30-.
R. Willemze et al. (1994) viste at forløpet av CD30+ lymfom er gunstigere. Genotypisk påvises klonal omorganisering av T-lymfocyttreseptoren.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
Symptomer T-celle-lymfomer i huden
Den vanligste sykdommen i gruppen av T-cellelymfomer i huden er mycosis fungoides, som står for omtrent 70 % av tilfellene. Det finnes tre kliniske former for sykdommen: klassisk, erytrodermisk og halshugget. T-cellelymfomer er karakterisert ved polymorfisme av utslett i form av flekker, plakk, svulster.
Den erytrodermiske formen av mycosis fungoides begynner vanligvis med ukontrollerbar kløe, hevelse, universell hyperemi, forekomst av erytematøse-plateepitellignende lesjoner på huden på overkroppen og ekstremitetene, som har en tendens til å smelte sammen og utvikle erytrodermi innen 1-2 måneder. Nesten alle pasienter har palmar-plantar hyperkeratose og diffus tynning av hår over hele huden. Alle grupper av lymfeknuter er sterkt forstørret. Forstørrede inguinale, femorale, aksillære, cubitale lymfeknuter palperes som "pakker" med tett elastisk konsistens, ikke smeltet sammen med omkringliggende vev, smertefrie. Den generelle tilstanden forverres kraftig: feber med en kroppstemperatur på opptil 38-39 ° C, nattesvette, svakhet og vekttap forekommer. For tiden anses Sezary syndrom av mange hudleger for å være den sjeldneste leukemiske varianten av den erytrodermiske formen av mycosis fungoides,
En uttalt leukocytose observeres i lymfocytogrammer - Sezary-celler. Sezary-celler er ondartede T-hjelpere, hvis kjerner har en foldet cerebriform overflate med dype invaginasjoner i kjernemembranen. Et dødelig utfall observeres etter 2-5 år, og den hyppige årsaken til dette er kardiovaskulær patologi og forgiftning.
Den halshuggede formen av mycosis fungoides er karakterisert ved rask utvikling av tumorlignende lesjoner på tilsynelatende frisk hud uten tidligere langvarig plakkdannelse. Denne formen er karakterisert ved en høy grad av malignitet, som regnes som en manifestasjon av lymfosarkom. Et dødelig utfall observeres innen et år.
Stages
Den klassiske formen for mycosis fungoides er preget av tre utviklingsstadier: erytematøs-plateepitel, plakk og tumor.
Det første stadiet ligner det kliniske bildet av noen godartede inflammatoriske dermatoser - eksem, seboreisk dermatitt, plakkparapsoriasis. På dette stadiet av sykdommen observeres flekker i forskjellige størrelser, intenst rosa, rosa-røde med et lilla skjær, runde eller ovale konturer, med relativt klare grenser, overfladisk kli-lignende eller finplate-avskalling. Elementene er ofte plassert på forskjellige områder av huden, oftest på overkroppen og ansiktet. Gradvis øker antallet. Over tid kan prosessen få karakter av erytrodermi (erytrodermisk stadium). Utslettet kan vare i årevis eller forsvinne spontant. I motsetning til godartede inflammatoriske dermatoser, er elementene i utslettet og kløen på dette stadiet resistente mot behandlingen.
Infiltrativt plakkstadium utvikler seg over flere år. I stedet for tidligere eksisterende flekkete utslett, oppstår plakk med runde eller uregelmessige konturer, intenst lilla i fargen, tydelig avgrenset fra sunn hud, tette, med en flassende overflate. Konsistensen ligner "tykk papp". Noen av dem forsvinner spontant og etterlater områder med mørkebrun hyperpigmentering og/eller atrofi (poikiloderma). Kløe på dette stadiet er enda mer intens og smertefull, feber og vekttap observeres. Lymfadenopati kan observeres på dette stadiet.
I det tredje, svulststadiet, oppstår smertefrie svulster med tett, elastisk konsistens, gulrød farge, som utvikler seg fra plakk eller oppstår på tilsynelatende sunn hud. Formen på svulstene er sfærisk eller flat, ofte som en sopphette. Svulster kan oppstå hvor som helst. Antallet varierer mye fra enkeltstående til dusinvis, størrelsen er fra 1 til 20 cm i diameter. Når lenge eksisterende svulster går i oppløsning, dannes det sår med ujevne kanter og dyp bunn som når fascia eller bein. Lymfeknuter, milt, lever og lunger påvirkes oftest. Den generelle tilstanden forverres, beruselsessymptomer oppstår og øker, og svakhet utvikler seg. Gjennomsnittlig levealder for pasienter med den klassiske formen for mycosis fungoides fra diagnosetidspunktet er fra 5 til 10 år. Dødelighet observeres vanligvis fra tilstøtende sykdommer: lungebetennelse, hjerte- og karsvikt, amyloidose. Subjektivt kjennes kløe, og når svulster går i oppløsning, smerter i de berørte områdene.
Hva trenger å undersøke?
Hvordan undersøke?
Behandling T-celle-lymfomer i huden
I det erytematøse-plateepitelstadiet trenger ikke pasienter antitumorbehandling; de får foreskrevet topiske kortikosteroider (prednisolon, betametason, deksametasonderivater), interferon alfa (3 millioner IE daglig, deretter 3 ganger i uken i 3-6 måneder, avhengig av kliniske manifestasjoner eller behandlingseffektivitet), interferon gamma (100 000 IE per dag i 10 dager, syklusen gjentas 12-3 ganger med en 10-dagers pause), PUVA-terapi eller Re-PUVA-terapi. Effektiviteten av PUVA-terapi er basert på selektiv dannelse av kovalente tverrbindinger av psoralener med DNA i prolifererende T-hjelperceller, noe som hemmer deres deling. I det andre stadiet brukes systemiske kortikosteroider (30-40 mg prednisolon per dag i 1,5-2 måneder) og cytostatika (prospedin 100 mg per dag daglig, totalt 4-5 injeksjoner) i tillegg til de ovennevnte midlene. Kombinasjon av interferoner med andre behandlingsmetoder har en mer uttalt terapeutisk effekt (interferoner + PUVA, interferoner + cytostatika, interferoner + aromatiske retinoider).
I tumorstadiet er hovedmetoden polykjemoterapi. En kombinasjon av vinkristin (0,5–1 mg intravenøst én gang daglig, totalt 4–5 injeksjoner) med prednisolon (40–60 mg per dag oralt under cellegiftbehandling), prospidin (100 mg per dag, totalt 3 g) og interferoner brukes. Fotodynamisk behandling, elektronstrålebehandling og fotoferese (ekstrakorporeal fotokjemoterapi) anbefales.