Medisinsk ekspert av artikkelen

Nye publikasjoner
Subakutt nekrotiserende Leahs encefalomyopati
Sist anmeldt: 04.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
 [
[ Fører til av Leahs syndrom
Sykdommen er basert på en mangel på enzymer som sørger for energiproduksjon, hovedsakelig på grunn av en forstyrrelse i pyruvinsyremetabolismen og en defekt i elektrontransport i respirasjonskjeden. Det utvikles en mangel på pyruvatdehydrogenasekomplekset (a-E1-subenhet), pyruvatkarboksylase, kompleks 1 (NAD-koenzym Q-reduktase) og kompleks 4 (cytokromoksidase) i respirasjonskjeden.
Det er fastslått at defekter i pyruvatkarboksylase, kompleks 1 (NAD-koenzym Q-reduktase) og kompleks 4 (cytokromoksidase) i respirasjonskjeden arves autosomalt recessivt, mens defekter i pyruvatdehydrogenasekomplekset (a-E1-subenhet) arves X-bundet recessivt. Ved punktmutasjoner i mtDNA, som påvirker den 6. subenheten av ATPase, er mitokondriell arv typisk. Oftest forekommer en miscens-mutasjon, assosiert med erstatning av tymin med guanin eller cytosin i posisjon 8993 av mtDNA. Mindre vanlig er en mutasjon i posisjon 9176 av mtDNA. Fordi T8993G-mutasjonen er hoveddefekten i NARP-syndrom, er familier med disse to sykdommene beskrevet. Hos barn er det også beskrevet en mutasjon i mtDNA i posisjon 8344, som forekommer ved MERRF-syndrom.
Det antas at ved akkumulering av mutant mtDNA i de fleste mitokondrier utvikles et alvorlig forløp av Leigh syndrom. Ved mitokondriedannelsen av denne tilstanden finnes mutant mtDNA i 90 % av alle mitokondrier. Patogenesen er assosiert med et brudd på energidannelsen i cellene og utviklingen av melkesyreacidose.
Symptomer av Leahs syndrom
De første tegnene på sykdommen debuterer i tidlig alder (1-3 år). Det er imidlertid kjente tilfeller av sykdomsmanifestasjon ved 2 uker og ved 6-7 års alder. Først utvikles uspesifikke lidelser: forsinket psykomotorisk utvikling, redusert appetitt, episoder med oppkast, vekttap. Deretter øker nevrologiske symptomer: muskelhypotoni eller dystoni med overgang til hypertoni, myokloniske anfall eller tonisk-kloniske anfall, skjelving i ekstremitetene, koreoatetose, koordinasjonsforstyrrelser, reduserte senereflekser, sløvhet, døsighet. Cerebral nevrodegenerasjon er progressiv. Symptomer på pyramidal og ekstrapyramidal insuffisiens øker, svelgeevnen svekkes. Slike endringer i synsorganet som ptose, oftalmoplegi, atrofi av synsnervene, sjeldnere pigmentdegenerasjon av netthinnen observeres ofte. Noen ganger utvikles hypertrofisk kardiomyopati, episoder med takypné oppstår.
I sjeldne tilfeller utvikler sykdommen seg som akutt encefalopati. Mer typisk er et kronisk eller subakutt forløp, som fører til dødelig utgang flere år etter sykdomsdebut. Ved et raskt forløp (flere uker) inntreffer døden som følge av lammelse av respirasjonssenteret.
Diagnostikk av Leahs syndrom
En biokjemisk blodprøve avslører melkesyreacidose på grunn av akkumulering av melkesyre og pyrodruesyre i blodet og cerebrospinalvæsken, samt en økning i alanininnholdet i blodet. Nivået av ketonlegemer kan også være forhøyet. Økt utskillelse av organiske syrer oppdages i urinen: melkesyre, fumarsyre, etc. Nivået av karnitin i blodet og vevene synker ofte.
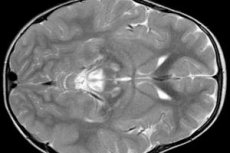
EEG-resultater avslører fokale tegn på epileptisk aktivitet. MR-data viser forstørrelse av hjerneventriklene, bilateral hjerneskade, forkalkning av basalgangliene (nucleus caudatus, putamen, substantia nigra, globus pallidus). Atrofi av hjernehalvdelene og hjernemassen kan også påvises.
Morfologisk undersøkelse avslører grove forandringer i hjernemassen: symmetriske nekrosefokus, demyelinisering og svampaktig degenerasjon av hjernen, hovedsakelig i de midtre delene, pons, basalgangliene, thalamus og synsnerven. Det histologiske bildet inkluderer cystisk degenerasjon av hjernevev, astrocytisk gliose, nevrondød og en økning i antall mitokondrier i cellene. I skjelettmuskulatur er det akkumulering av lipidinneslutninger, en reduksjon i den histokjemiske reaksjonen på kompleksene 1 og 4 i respirasjonskjeden, subsarkolemmal akkumulering av mitokondrier, unormale mitokondrier med desorganisering av cristae. RRF-fenomenet oppdages ofte ikke.
Hvordan undersøke?
Hvilke tester er nødvendig?
Использованная литература