Medisinsk ekspert av artikkelen

Nye publikasjoner
Polymyositt og dermatomyositt: årsaker, symptomer, diagnose, behandling
Sist anmeldt: 05.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
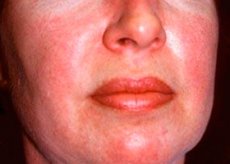
Polymyositt og dermatomyositt er sjeldne systemiske revmatiske sykdommer som kjennetegnes av inflammatoriske og degenerative forandringer i muskler (polymyositt) eller muskler og hud (dermatomyositt). Den mest spesifikke hudmanifestasjonen er heliotroputslett.
Muskelaffeksjon er symmetrisk og inkluderer svakhet, noe ømhet og påfølgende atrofi av de proksimale bekkenbeltemusklene. Komplikasjoner kan inkludere visceral affeksjon og malignitet. Diagnosen er basert på klinisk presentasjon og vurdering av muskeldysfunksjon ved å måle enzymnivåer, utføre MR, elektromyografi og muskelbiopsi. Behandlingen involverer glukokortikoider, noen ganger i kombinasjon med immunsuppressiva eller intravenøse immunglobuliner.
Kvinner blir dobbelt så ofte som menn. Sykdommen kan oppstå i alle aldre, men oppdages oftest i alderen 40 til 60 år; hos barn - fra 5 til 15 år.
 [
[ Hva forårsaker dermatomyositt og polymyositt?
Årsaken til sykdommen antas å være en autoimmun reaksjon på muskelvev hos genetisk disponerte individer. Sykdommen er mer vanlig ved en belastet familiehistorie og bærere av visse HLA-antigener (DR3, DR52, DR56). Mulige utløsere er viral myositt og ondartede neoplasmer. Det finnes rapporter om påvisning av strukturer som ligner på picornavirus i muskelceller; i tillegg kan virus indusere lignende sykdommer hos dyr. Sammenhengen mellom ondartede svulster og dermatomyositt (mye sjeldnere enn med polymyositt) antyder at tumorvekst også kan være en utløser for utviklingen av sykdommen som et resultat av initiering av autoimmune reaksjoner på vanlige antigener i svulsten og muskelvevet.
Avleiringer av IgM, IgG og den tredje komponenten av komplement finnes i veggene i blodårene i skjelettmuskulaturen; dette er spesielt karakteristisk for dermatomyositt hos barn. Pasienter med polymyositt kan også utvikle andre autoimmune prosesser.
Patofysiologi av dermatomyositt og polymyositt
Patologiske forandringer inkluderer celleskade og atrofi mot bakgrunn av betennelse av varierende alvorlighetsgrad. Musklene i øvre og nedre ekstremiteter, samt ansiktet, er skadet i mindre grad enn andre skjelettmuskler. Skade på viscerale muskler i svelget og øvre spiserør, sjeldnere hjerte, mage eller tarm, kan føre til dysfunksjon av de ovennevnte organene. Høye konsentrasjoner av myoglobin på grunn av rabdomyolyse kan forårsake nyreskade. Inflammatoriske forandringer i ledd og lunger kan også forekomme, spesielt hos pasienter som har antisyntetase-antistoffer.
Symptomer på dermatomyositt og polymyositt
Polymyositt kan utvikle seg akutt (spesielt hos barn) eller subakutt (oftere hos voksne). Akutt virusinfeksjon går noen ganger forut for eller er en utløsende faktor for sykdommens manifestasjon, og de vanligste manifestasjonene er svakhet i proksimale muskler eller hudutslett. Smerte uttrykkes i mindre grad enn svakhet. Polyarthralgi, Raynauds fenomen, dysfagi, lungesykdommer, generelle symptomer (økt kroppstemperatur, redusert kroppsvekt, svakhet) kan utvikle seg. Raynauds fenomen finnes ofte hos pasienter med samtidig bindevevssykdommer.
Muskelsvakhet kan utvikle seg over flere uker eller måneder. For at muskelsvakhet skal manifestere seg klinisk, må imidlertid minst 50 % av muskelfibrene være påvirket (dermed indikerer tilstedeværelsen av muskelsvakhet progresjon av myositt). Pasienter kan oppleve problemer med å heve armene over skulderhøyde, gå i trapper eller reise seg fra sittende stilling. På grunn av alvorlig svakhet i bekken- og skulderbeltemusklene kan pasientene være bundet til rullestol eller seng. Hvis nakkebøyerne påvirkes, blir det umulig å løfte hodet fra puten. Påvirkning av musklene i svelget og øvre spiserør fører til svelgeproblemer og oppstøt. Musklene i nedre, øvre lemmer og ansiktet påvirkes vanligvis ikke. Imidlertid kan kontrakturer i lemmene utvikle seg.
Hudutslett observert ved dermatomyositt er vanligvis mørke i fargen og erytematøse. Periorbitalt ødem med lilla farge (heliotroputslett) er også karakteristisk. Hudutslett kan være litt forhøyet over hudnivået og være glatt eller skjellete; utslettet er lokalisert i panne, nakke, skuldre, bryst, rygg, underarmer, nedre deler av leggene, øyenbryn, kneområder, mediale malleoler, dorsale overflater av interfalangeale og metakarpofalangeale ledd, på sidesiden (Gottrons symptom). Hyperemi i bunnen eller periferien av neglene er mulig. Deskvamativ dermatitt, ledsaget av sprekker, kan utvikle seg på huden på sideflaten av fingrene. Primære hudlesjoner forsvinner ofte uten følgevirkninger, men kan føre til sekundære forandringer som mørk pigmentering, atrofi, arrdannelse eller vitiligo. Subkutan forkalkning kan utvikle seg, spesielt hos barn.
Omtrent 30 % av pasientene utvikler polyarthralgi eller polyartritt, ofte ledsaget av ødem og leddeffusjon. Alvorlighetsgraden av leddmanifestasjonene er imidlertid liten. De forekommer oftere når antistoffer mot Jo-1 eller andre syntetaser påvises hos pasientene.
Affeksjon av indre organer (unntatt svelget og øvre del av spiserøret) er mindre vanlig ved polymyositt enn ved andre revmatiske sykdommer (spesielt SLE og systemisk sklerose). I sjeldne tilfeller, spesielt ved antisyntetasesyndrom, manifesterer sykdommen seg som interstitiell pneumonitt (i form av dyspné og hoste). Hjerterytmeforstyrrelser og ledningsforstyrrelser kan utvikles, men de er vanligvis asymptomatiske. Gastrointestinale manifestasjoner er vanligere hos barn som også har vaskulitt og kan inkludere oppkast med blod, melena og tarmperforasjon.
Hvor gjør det vondt?
Klassifisering av polymyositt
Det finnes 5 typer polymyositt.
- Primær idiopatisk polymyositt, som kan oppstå i alle aldre, involverer ikke huden.
- Primær idiopatisk dermatomyositt ligner på primær idiopatisk polymyositt, men involverer huden.
- Polymyositt og dermatomyositt assosiert med ondartede neoplasmer kan forekomme hos pasienter i alle aldre; utviklingen av disse observeres oftest hos eldre pasienter, så vel som hos pasienter med andre bindevevssykdommer. Utviklingen av ondartede neoplasmer kan observeres både innen 2 år før og innen 2 år etter at myositten har oppstått.
- Polymyositt eller dermatomyositt hos barn er assosiert med systemisk vaskulitt.
- Polymyositt og dermatomyositt kan også forekomme hos pasienter med andre bindevevssykdommer, oftest progressiv systemisk sklerose, blandet bindevevssykdom og SLE.
Det er feil å inkludere myositt i truncalmuskulaturen i gruppen polymyositt, siden sistnevnte er en egen sykdom som er karakterisert av kliniske manifestasjoner som ligner på kronisk idiopatisk polymyositt. Den utvikler seg imidlertid i alderdommen, påvirker ofte musklene i kroppens distale deler (for eksempel øvre og nedre ekstremiteter), har lengre varighet, responderer dårligere på behandling og er preget av et typisk histologisk bilde.
Diagnose av dermatomyositt og polymyositt
Polymyositt bør mistenkes hos pasienter med klager over proksimal muskelsvakhet, med eller uten ømhet. Utredning for dermatomyositt er nødvendig hos pasienter med klager over utslett som ligner heliotrop eller Gottrons tegn, samt hos pasienter med manifestasjoner av polymyositt kombinert med hudlesjoner forenlige med dermatomyositt. De kliniske manifestasjonene av polymyositt og dermatomyositt kan ligne på de ved systemisk sklerose eller, sjeldnere, SLE eller vaskulitt. Sikkerheten i diagnosen økes ved å oppfylle så mange av følgende fem kriterier som mulig:
- svakhet i proksimale muskler;
- karakteristiske hudutslett;
- økt aktivitet av muskelvevsenzymer (kreatinkinase eller, i fravær av økning i aktiviteten, aminotransferaser eller aldolase);
- karakteristiske endringer i myografi eller MR;
- karakteristiske histologiske forandringer i en muskelvevsbiopsi (absolutt kriterium).
Muskelbiopsi kan utelukke noen klinisk lignende tilstander, som myositt i truncusmuskulaturen og rabdomyolyse på grunn av virusinfeksjon. Forandringer avdekket ved histologisk undersøkelse kan variere, men kronisk betennelse, fokus på muskeldegenerasjon og regenerering er typiske. En nøyaktig diagnose (vanligvis ved histologisk verifisering) er nødvendig før potensielt toksisk behandling startes. MR kan oppdage fokus på ødem og betennelse i musklene, etterfulgt av målrettet biopsi av disse.
Laboratoriestudier kan bekrefte eller tvert imot eliminere mistanken om sykdommens tilstedeværelse, og er også nyttige for å vurdere alvorlighetsgraden, muligheten for en kombinasjon med annen lignende patologi og diagnostisere komplikasjoner. Til tross for at antinukleære antistoffer oppdages hos noen pasienter, er dette fenomenet mer typisk for andre bindevevssykdommer. Omtrent 60 % av pasientene har antistoffer mot antigenet i kjerner (PM-1) eller hele tymusceller og Jo-1. Rollen til autoantistoffer i sykdommens patogenese er fortsatt uklar, selv om det er kjent at antistoffer mot Jo-1 er en spesifikk markør for antisyntetasesyndrom, inkludert fibroserende alveolitt, lungefibrose, leddgikt og Raynauds fenomen.
Periodisk vurdering av kreatinkinaseaktivitet er nyttig for å overvåke behandlingen. Hos pasienter med alvorlig muskelsvinn kan imidlertid enzymaktiviteten være normal til tross for kronisk aktiv myositt. MR, muskelbiopsi eller forhøyet kreatinkinaseaktivitet er ofte nyttige for å skille mellom tilbakefall av polymyositt og glukokortikoidindusert myopati.
Fordi mange pasienter har udiagnostiserte maligniteter, anbefaler noen forfattere screening av alle voksne med dermatomyositt og de med polymyositt over 60 år ved hjelp av følgende tidsplan: fysisk undersøkelse, inkludert bryst-, bekken- og endetarmsundersøkelser (inkludert avføringstesting for okkult blod); fullstendig blodtelling; blodkjemi; mammografi; karsinoembryonisk antigentest; urinanalyse; røntgen av brystet. Noen forfattere stiller spørsmål ved behovet for slik screening hos yngre pasienter som ikke har kliniske tegn på malignitet.
Hva trenger å undersøke?
Hvordan undersøke?
Behandling av dermatomyositt og polymyositt
Inntil betennelsen er lindret, bør fysisk aktivitet begrenses. Glukokortikoider er førstelinjemedisiner. I den akutte fasen av sykdommen bør voksne pasienter foreskrives prednisolon (oralt) i en dose på 40 til 60 mg per dag. Regelmessig bestemmelse av kreatinkinaseaktivitet er en tidlig indikator på effektivitet: hos de fleste pasienter observeres en reduksjon eller normalisering innen 6 til 12 uker etter en økning i muskelstyrke. Etter normalisering av enzymaktivitet reduseres prednisolondosen: først med omtrent 2,5 mg per dag over en uke, deretter raskere; hvis økt muskelenzymaktivitet kommer tilbake, økes hormondosen igjen. Tilfrisknede pasienter kan klare seg uten glukokortikoider, men oftest trenger voksne pasienter langvarig glukokortikoidbehandling (10-15 mg prednisolon per dag). Startdosen av prednisolon for barn er 30-60 mg/m² én gang daglig. Ved remisjon i > 1 år hos barn, kan glukokortikoidbehandling seponeres.
I noen tilfeller opplever pasienter som får høye doser glukokortikoider en plutselig økning i muskelsvakhet, noe som kan være forbundet med utviklingen av glukokortikoidmyopati.
Ved utilstrekkelig respons på glukokortikoidbehandling, samt ved utvikling av glukokortikoidmyopati eller andre komplikasjoner som krever dosereduksjon eller seponering av prednisolon, bør immunsuppressive midler (metotreksat, cyklofosfamid, azatioprin, ciklosporin) brukes. Noen pasienter kan få kun metotreksat (vanligvis i doser som overstiger de som brukes ved behandling av RA) i mer enn 5 år. Intravenøse immunglobuliner kan være effektive hos pasienter som er refraktære mot medikamentell behandling, men bruken av disse øker behandlingskostnadene.
Myositt assosiert med primære og metastatiske svulster, samt myositt i stammemuskulaturen, er vanligvis mer refraktære mot glukokortikoidbehandling. Remisjon av myositt assosiert med ondartede svulster er mulig etter fjerning av svulsten.
Hva er prognosen for dermatomyositt og polymyositt?
Langvarig remisjon (og til og med klinisk bedring) over 5 år observeres hos mer enn halvparten av behandlede pasienter; hos barn er dette tallet høyere. Tilbakefall kan imidlertid forekomme når som helst. Den totale femårsoverlevelsesraten er 75 %, høyere hos barn. Dødsårsakene hos voksne er alvorlig og progressiv muskelsvakhet, dysfagi, redusert ernæring, aspirasjonspneumoni eller respirasjonssvikt på grunn av lungeinfeksjoner. Polymyositt er mer alvorlig og resistent mot behandling hvis det er skade på hjerte og lunger. Død hos barn kan oppstå på grunn av intestinal vaskulitt. Den generelle prognosen for sykdommen bestemmes også av tilstedeværelsen av ondartede neoplasmer.