Medisinsk ekspert av artikkelen

Nye publikasjoner
Angelmans syndrom hos barn og voksne
Sist anmeldt: 04.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.

Det finnes en rekke sykdommer der uttrykk som «ta vare på deg selv, så blir du ikke syk» høres minst latterlige ut. Dette er patologier der noen mentale og fysiske abnormiteter er iboende i barnets kropp allerede før fødselen, men foreldrene har ikke skylden for dette. Slike sykdommer er forårsaket av mutasjoner eller abnormiteter i kromosomsett og kalles kromosomale eller genetiske. Angelman syndrom, Downs syndrom, Patau syndrom, Edwards syndrom, Turners syndrom, Prader-Willis syndrom - dette er bare en del av de genetiske sykdommene fra en ganske anstendig liste.
Lykkemannssyndrom
Denne gangen skal vi snakke om patologien oppkalt etter den engelske barnelegen Harry Angelman, som først tok opp spørsmålet om dette problemet i 1965, etter å ha møtt tre uvanlige barn i sin praksis dagen før, forent av felles særegne symptomer. Legen kalte disse barna dukkebarn og skrev en artikkel om dem, som opprinnelig het «Barn-marionetter». Selve artikkelen og tittelen ble skrevet under inntrykk av et maleri sett i et av museene i Verona. Maleriet avbildet en leende gutt, og det ble kalt «Dukkegutten». Assosiasjonen mellom barnet som er avbildet i maleriet og de tre barna som Angelman en gang møtte i sin praksis, fikk barnelegen til å slå sammen barna til én gruppe på grunn av sykdommen de hadde.
Det er ikke noe overraskende i det faktum at barna som er nevnt i artikkelen ikke ble lagt merke til av andre leger. Tross alt virket det ved første øyekast som om de hadde helt forskjellige sykdommer, så forskjellig var det generelle kliniske bildet av sykdommen i 3 forskjellige tilfeller. Kanskje den "nye" kromosomale patologien ville ha interessert andre forskere, men på den tiden var genetikken ennå ikke utviklet nok til å bekrefte hypotesen til den engelske legen. Derfor, etter en viss interesse for den, ble artikkelen kastet på bakre hylle i lang tid.
Den neste omtalen av Angelman syndrom, som er det artikkelen av den engelske barnelegen G. Angelman nå ble kalt, stammer fra begynnelsen av 80-tallet av 1900-tallet. Og først i 1987 var det mulig å finne årsaken til at en liten del av barn blir født med slike avvik at de utenfra ser ut til å være konstant smilende og glade. Faktisk er dette slett ikke sant, og smilet er bare en grimase, bak hvilken det skjuler seg en ulykkelig menneskesjel og foreldrenes smerte.
Epidemiologi
Ifølge statistikk kan en kromosommutasjon hos et barn utvikle seg både mot bakgrunn av lignende mutasjoner hos foreldre og i fravær av slike. Det finnes ingen klar arvelig natur ved Angelman syndrom (AS), men sannsynligheten for å utvikle patologi hos foreldre med kromosomale mutasjoner er ganske høy.
Det er også interessant at hvis en familie allerede har et barn med AS, er det én prosent sjanse for å få et andre barn med samme lidelse, selv om foreldrene er friske.
Det finnes fortsatt ingen eksakt statistikk over antall pasienter med Angelmans syndrom. Kanskje årsaken er variasjonen i symptomene, som kan forekomme i en viss sammensetning eller ikke forekomme i det hele tatt over lengre tid. Det antas at sykdommens forekomst er: 1 barn per 20 000 nyfødte. Men dette tallet er svært omtrentlig.
Fører til Angelmans syndrom
Angelmans syndrom er et medisinsk navn for en kromosomal patologi, men det er langt fra det eneste. Folk kaller denne sykdommen dukkebarnssyndrom, gladdukkesyndrom, Petrusjka-syndrom og latterdukkesyndrom. Folk kommer opp med alle slags navn (noen ganger til og med støtende for pasientene selv og foreldrene deres), men en sykdom er en sykdom, uansett hvor morsom den kan se ut og uansett hva årsakene er.
Og årsakene til utviklingen av Angelman syndrom, som mange andre genetiske patologier, er i alle tilfeller forstyrrelser i strukturen til ett av kromosomene eller kromosomsettet som helhet. Men i vårt tilfelle ligger hele problemet i kromosom 15, som er arvet fra moren. Det vil si at farskromosomet i dette tilfellet ikke har noen avvik, men det kvinnelige gjennomgår visse mutasjoner.
I henhold til typen kromosomavvik klassifiseres Angelmans syndrom som en kromosomal mutasjon. Slike mutasjoner anses å være:
- En delesjon (fravær av en del av et kromosom som inneholder et visst sett med gener; hvis ett av genene mangler, snakker vi om en mikrodelesjon), som er resultatet av to brudd og én gjenforening, når en del av det opprinnelige kromosomet går tapt.
- Duplisering (tilstedeværelsen av en ekstra seksjon i et kromosom som er en kopi av en eksisterende), som i de fleste tilfeller fører til en persons død, og sjeldnere til infertilitet.
- Inversjon (reversering av en av kromosomseksjonene med 180 grader, dvs. i motsatt retning, og så er genene i den plassert i motsatt rekkefølge), når de ødelagte endene av kromosomet er koblet sammen i en annen rekkefølge enn originalen.
- Innsetting (hvis deler av det genetiske materialet i et kromosom er ute av plass),
- translokasjon (hvis en viss del av et kromosom er festet til et annet kromosom; en slik mutasjon kan være gjensidig uten tap av seksjoner).
Hvis barnet mottar et mutert kromosom fra en intetanende mor, er det dømt til å bli født med abnormaliteter. Den vanligste årsaken til Angelmans syndrom anses fortsatt å være en sletting av morens 15. kromosom, når en liten del mangler. Mindre vanlige mutasjoner i "latterdukke"-syndromet anses å være:
- translokasjon,
- unipaternal disomi (hvis barnet fikk et par kromosomer fra faren, er morskromosomet fraværende),
- mutasjon av gener i DNA, som både er hovedbyggematerialet (genetisk materiale) og instruksjoner for riktig bruk (spesielt mutasjon av ube3a-genet i mors kromosom).
Tilstedeværelsen av en av disse mutasjonene hos foreldre er en risikofaktor for utvikling av Angelmans syndrom hos barn. Men ikke bare kromosomale mutasjoner, men også genomiske (som er assosiert med en kvantitativ endring i kromosomsett og er vanligere enn kromosomale) kan provosere frem utviklingen av sykdommen hos et barn. Vanlige genomiske mutasjoner inkluderer kromosomtrisomi (hvis en persons kromosomsett har mer enn 46 kromosomer).
For at en patologi skal oppstå hos et barn, er det slett ikke nødvendig at foreldrene har kromosomavvik. Likevel er det en viss prosentandel av pasienter hvis sykdom er arvelig.
Patogenesen
La oss dykke litt dypere inn i biologi, eller mer presist, genetikk. Den genetiske informasjonen til hver enkelt menneskelig organisme finnes i 23 kromosompar. Ett kromosom fra et par overføres til barnet fra faren, det andre fra moren. Alle kromosomparene er forskjellige i form og størrelse og bærer spesifikk informasjon. Dermed er det 23. kromosomparet (X- og Y-kromosomer) ansvarlig for dannelsen av babyens seksuelle egenskaper (XX - jente, XY - gutt, mens Y-kromosomet bare kan mottas av barnet fra faren).
Ideelt sett får et barn 46 kromosomer fra foreldrene sine, som danner barnets genetiske egenskaper og forhåndsbestemmer det som individ. Et større antall kromosomer kalles trisomi og regnes som et avvik fra normen. For eksempel forårsaker tilstedeværelsen av kromosom 47 i kromosomsettet (karyotype, artsbestemmende og individuelle egenskaper) forekomsten av Downs syndrom.
Hvis kromosomene er farget med et spesielt fargestoff, kan man under mikroskopet se striper i forskjellige nyanser langs hver av dem. Inne i hver stripe er det et stort antall gener. Alle disse stripene er nummerert av forskere og har en fast plassering. Fraværet av en av stripene regnes som et avvik fra normen. Ved Angelman syndrom kan man ofte observere fravær av segmenter av morskromosomet i intervallet q11-q13, plassert i den lange armen, hvor antallet DNA-baser bare er omtrent 4 millioner.
Hovedkomponenten i kromosomet regnes som et utrolig langt DNA-molekyl som inneholder tusenvis av gener og titalls og hundrevis av millioner nitrogenholdige baser. Dermed inneholder kromosom 15, som er ansvarlig for utviklingen av Angelman syndrom og flere andre, 1200 gener og omtrent 100 millioner baser. Eventuelle forstyrrelser i DNA-molekylets struktur vil garantert påvirke det fremtidige barnets utseende og utvikling.
Den genetiske informasjonen i gener omdannes til protein eller RNA. Denne prosessen kalles genuttrykk. På denne måten får den genetiske informasjonen som mottas fra foreldrene både form og innhold, som er nedfelt i deres unike kvinnelige eller mannlige arving.
Det finnes en rekke patologier med en ikke-klassisk type arv, inkludert Angelman syndrom, der gener mottatt fra foreldre som en del av parede kromosomer bærer et unikt preg av foreldrene og manifesterer seg på forskjellige måter.
Så, Angelman syndrom er et slående eksempel på genomisk imprinting, der genuttrykk i barnets kropp er direkte avhengig av hvilken forelder allelene ble mottatt fra (forskjellige former av ett gen, mottatt fra far og mor, plassert på identiske deler av parede kromosomer). Det vil si at bare anomalier i morskromosomet fører til utviklingen av syndromet, mens mutasjoner og strukturelle forstyrrelser i farskromosomet forårsaker helt forskjellige patologier.
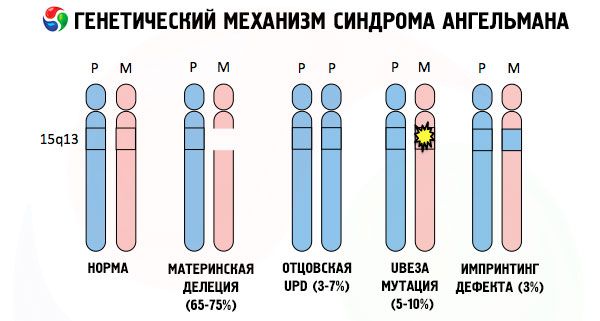
Ved denne patologien er det mangel på visse gener i morens kromosom eller tap/reduksjon i aktiviteten til individuelle gener (i de aller fleste tilfeller ube3a-genet, som er involvert i metabolismen av ubiquitin, et protein som regulerer nedbrytningen av andre proteiner). Som et resultat av dette får barnet diagnosen psykiske utviklingsavvik og fysiske deformiteter.
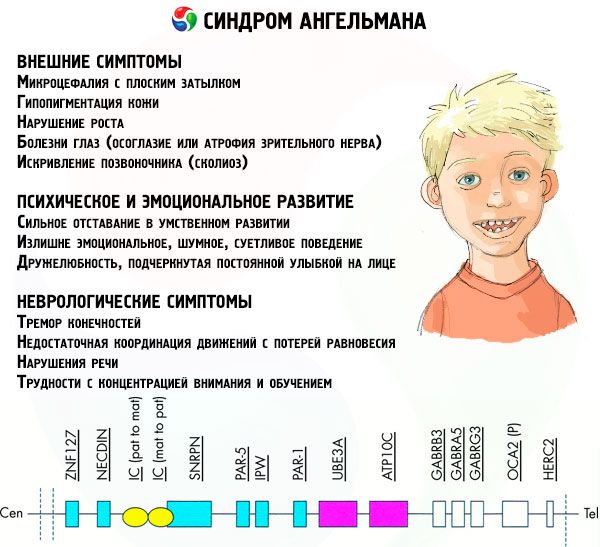
Symptomer Angelmans syndrom
Symptomene på Angelman syndrom påvirker ulike aspekter av et barns liv og utvikling: fysiske, nevrologiske, mentale. Basert på dette kan man identifisere tre grupper av symptomer som indikerer utviklingen av denne patologien.
- Eksterne eller fysiske symptomer:
- et uforholdsmessig lite hode sammenlignet med kroppen og lemmene, som er av normal størrelse,
- for bred munn,
- det er nesten alltid et smil om munnen (med åpen munn),
- sparsomme tenner,
- smal overleppe,
- ofte utstående bred tunge,
- utstående underkjeven,
- spiss hake,
- veldig lys hud, ofte hår (albinisme, assosiert med at kroppen ikke produserer pigmentet melanin),
- mørke flekker på lys hud (hypopigmentering på grunn av utilstrekkelig melaninproduksjon)
- fysiske eller ytre symptomer: øyesykdommer som strabismus eller atrofi av synsnerven,
- krumning av ryggraden (skoliose),
- stive ben (når man går, bøyer ikke en person bena i knærne på grunn av lav mobilitet i leddene, derav sammenligningen med en dukkegang).
- Symptomer relatert til mental og emosjonell utvikling:
- alvorlig psykisk utviklingshemming,
- overdrevent emosjonell, støyende, masete oppførsel,
- hyppig klapping i hendene,
- uttrykte vennlighet, understreket av et konstant smil om munnen,
- hyppig latter uten grunn.
- Nevrologiske symptomer:
- skjelving i lemmene,
- utilstrekkelig koordinering av bevegelser med tap av balanse,
- redusert muskeltonus,
- ulike søvnforstyrrelser,
- hyppige hysteriske anfall i barndommen,
- taleforstyrrelser (barnet begynner å snakke sent, har dårlige kommunikasjonsevner og utydelig tale),
- hyperaktivitet mot bakgrunn av økt eksitabilitet,
- vansker med å konsentrere seg og lære.
Men dette er et generalisert bilde av sykdommen. Faktisk avhenger det kliniske bildet av Angelman syndrom i stor grad av sykdommens utviklingsstadium og typen kromosommutasjon som forårsaket patologien. Dette betyr at symptomene på sykdommen kan variere betydelig hos forskjellige pasienter, noe som lenge ikke tillot oss å skille patologien fra andre med et lignende klinisk bilde.
Blant det totale antallet symptomer kan vi fremheve de som er karakteristiske for alle pasienter uten unntak:
- alvorlig psykisk utviklingshemming,
- upassende oppførsel (urimelig latter, økt opphisselse, dårlig konsentrasjon, eufori),
- underutvikling av motoriske ferdigheter,
- dårlig koordinasjon av bevegelser, gangart ataksi (ujevn tempo, svaiing fra side til side, etc.), skjelving i lemmene.
- taleutviklingsforstyrrelse med overvekt av ikke-verbale kommunikasjonsmidler.
Blant symptomene som de aller fleste pasienter opplever, kan følgende skilles ut:
- uforhold mellom hode og kropp forårsaket av forsinket fysisk utvikling,
- hos mange pasienter er hodeskallens form slik at hjernens størrelse forblir mindre enn hos friske mennesker (mikrocefali),
- epileptiske anfall før 3-årsalderen med en progressiv reduksjon i styrke og hyppighet ved høyere alder,
- forvrengning av EEG-parametere (fluktuasjoner og høy amplitude av lavfrekvente bølger).
Disse symptomene er ganske vanlige, men 20 % av pasientene med Angelmans syndrom har dem ikke.
Enda sjeldnere er det mulig å diagnostisere slike manifestasjoner av sykdommen som:
- alvorlig eller mild strabismus,
- dårlig kontroll over tungebevegelsen, noe som resulterer i at pasienter ofte stikker ut tungen uten grunn,
- vansker med å svelge og suge, spesielt hos små barn,
- forstyrrelse av hud- og øyepigmentering,
- armene hevet eller bøyd mens man går,
- hyperrefleksi,
- søvnforstyrrelser, spesielt i barndommen,
- hyppig spyttsekresjon,
- umettelig tørst,
- overaktive tyggebevegelser,
- overfølsomhet for varme,
- flat bakside av hodet,
- utstående underkjeven,
- glatte håndflater.
En ganske stor andel av pasientene har problemer med vannlating, som de har dårlig kontroll over, svekket finmotorikk, noe som skaper vansker med egenomsorg og læring, og overvekt. Nesten alle pasienter opplever puberteten senere enn friske jevnaldrende.
Barn med Angelman syndrom oppfatter muntlig tale godt og forstår den, men ønsker ikke å delta i samtaler, og begrenser derfor talen til flere dusin ord som er nødvendige i hverdagen. I voksen alder ser imidlertid slike pasienter yngre ut enn sine jevnaldrende uten genetiske patologier.
Mange symptomer på Angelmans syndrom er inkonstante, så det kliniske bildet av sykdommen endrer seg betydelig med alderen. Krampeanfall og epileptiske anfall blir sjeldnere eller forsvinner helt, pasienten blir mindre opphisset, og søvnen forbedres.
Komplikasjoner og konsekvenser
Angelmans syndrom er en alvorlig, for tiden så godt som uhelbredelig kromosompatologi som fratar pasienter muligheten til å leve et normalt liv. Hvordan livet til et barn med AS vil være, avhenger i stor grad av typen kromosomavvik.
Duplisering av et kromosomsegment er uforenlig med liv i de fleste tilfeller. Og selv om slike pasienter ikke dør i spedbarnsalderen og når puberteten, har de ingen sjanse til å få barn.
Sletting eller fravær av en del av genene som forekommer oftest ved Angelmans syndrom er et hinder for at barnet skal lære å gå og snakke. Slike barn har en mer alvorlig form for psykisk utviklingshemming, og epileptiske anfall forekommer oftere, og intensiteten deres er mye større enn hos pasienter med andre kromosomavvik.
Hvis det bare er en mutasjon av ett gen, kan barnet med tilstrekkelig oppmerksomhet og tilnærming lære det grunnleggende om egenomsorg, kommunikasjon og samhandling i en gruppe, selv om det fortsatt vil henge etter sine jevnaldrende i utvikling.
For barn med Angelman syndrom, som er snille av natur, er det viktigste foreldrenes kjærlighet og oppmerksomhet. Bare i dette tilfellet vil barnets utdanning bære frukter, selv om den er liten. Pasienter med AS vil selvfølgelig ikke kunne studere på en vanlig skole. De trenger spesialklasser der barn først lærer å konsentrere seg, og deretter gradvis gis grunnleggende skolekunnskaper.
Diagnostikk Angelmans syndrom
Angelmans syndrom er en medfødt utviklingspatologi. Men på grunn av visse omstendigheter er det ofte umulig å diagnostisere det i spedbarnsalderen og tidlig barndom. Dette skyldes manglende spesifisitet og svakt uttrykk av symptomer hos spedbarn og barn under 3 år. Og sykdommens forekomst i vårt land er ikke så stor at leger har lært å gjenkjenne den blant jevnaldrende.
Angelmans syndrom hos spedbarn kan manifestere seg som redusert muskeltonus, som manifesterer seg i problemer med mating (svakhet i suge- og svelgerefleksen), og senere vansker med å lære å gå (slike barn begynner å gå mye senere). Disse symptomene er de første tegnene på en utviklingsforstyrrelse hos babyen, som godt kan være assosiert med en kromosomavvik. Bare genetisk analyse kan bekrefte denne antagelsen.
Spesiell oppmerksomhet rettes mot barn hvis foreldre har ulike genomiske eller kromosomale lidelser. Tross alt kan sykdommen ikke manifestere seg i starten, og hvis patologien oppdages i tide, er det mulig å oppnå betydelig større suksess i læring ved å begynne å jobbe intensivt med barnet, noe som bremser sykdomsprogresjonen.
Hvis foreldrene har ulike kromosomavvik, utføres genetisk analyse selv før babyen er født, siden SA er en av patologiene som kan oppdages i embryonalstadiet.
Innsamling av materiale til genetisk forskning kan utføres på to måter:
- invasiv (med en viss prosentandel av risiko, siden det er nødvendig å trenge inn i livmoren for å ta en prøve av fostervann),
- ikke-invasiv (analyse av babyens DNA fra morens blod).
Følgende studier utføres deretter:
- fluorescerende in situ-hybridisering (FISH-metoden) – binding av en DNA-probe merket med et spesielt fargestoff til DNA-et som studeres, etterfulgt av undersøkelse under et mikroskop.
- analyse av mutasjoner i ube3a-genet og imprintede gener,
- DNA-metyleringsanalyse ved bruk av spesielle metoder brukt i genetikk.
Genetiske tester gir ganske nøyaktig informasjon ved kromosomavvik, noe som betyr at vordende foreldre vet på forhånd hva de skal forberede seg på. Det finnes imidlertid unntak. Hos en viss pasientgruppe, dersom alle symptomene som indikerer patologi er til stede, forblir testresultatene normale. Det vil si at patologi bare kan identifiseres ved å nøye observere barnet fra tidlig barndom: hvordan det spiser, når det begynte å gå og snakke, om det bøyer beina når det går, osv.
I tillegg til FISH-metoden kan man blant de instrumentelle diagnostiske metodene for Angelman syndrom skille ut tomografi (CT eller MR), som bidrar til å bestemme hjernens tilstand og størrelse, og et elektroencefalogram (EEG), som viser hvordan individuelle deler av hjernen fungerer.
Leger stiller vanligvis en endelig diagnose i alderen 3–7 år, når pasienten allerede har de fleste symptomene og dynamikken i sykdomsutviklingen er synlig.
Hvilke tester er nødvendig?
Differensiell diagnose
Angelmans syndrom er en genetisk patologi som praktisk talt ikke har noen spesifikke manifestasjoner. De fleste symptomene kan indikere både AS og andre genetiske patologier.
Differensialdiagnose av Angelman syndrom utføres med følgende patologier:
- Pitt-Hopkins syndrom (pasientene er preget av psykisk utviklingshemming, munter natur, smilende, de har en ganske stor og bred munn, mikrocefali er notert). Forskjellen er anfall av hyperventilering og pusteholding i våken tilstand.
- Christiansons syndrom (pasientene er psykisk utviklingshemmede personer med munter gemytt, ute av stand til å snakke, preget av mikrocefali, ataksi, kramper, ufrivillige muskelbevegelser).
- Mowat-Wilsons syndrom (symptomer: psykisk utviklingshemming, epileptiske anfall, spiss hake, åpen munn, glad ansiktsuttrykk, mikrocefali). Kjennetegn: stor avstand mellom øynene, øynene skrått innover, avrundet nesetipp, bakovervendt øremuskel.
- Kabuki-syndrom (karakterisert av mild til moderat psykisk utviklingshemming, tale- og motoriske problemer, muskelsvakhet, epileptiske anfall, mikrocefali, lange kløeintervaller og nedsatt koordinasjon). Karakterisert av buede øyenbryn, utadvendt lateral del av det nedre øyelokket, vidt plasserte øyne, lange palpebralfissurer med lange, tykke øyevipper.
- Rett syndrom (differensiering fra AS hos kvinner). Symptomer: forsinket taleutvikling, anfall, mikrocefali. Forskjellen er at det ikke er noe lykkelig uttrykk i ansiktet, det er anfall av apné og apraksi, som utvikler seg over tid.
- Autosomalt recessivt mentalt tardasjonssyndrom 38 (symptomer: markert mental retardasjon med forsinkelser i motoriske ferdigheter og tale, muskelsvakhet, spiseproblemer i spedbarnsalderen, impulsivitet). Kjennetegn er irisens blå farge.
- MECP 2-genduplikasjonssyndrom (differensiering fra SA hos menn). Symptomer: alvorlig psykisk utviklingshemming, muskelsvakhet siden barndommen, taleproblemer eller mangel på tale, epilepsi. Kjennetegn: progressiv myopati, stadig tilbakevendende infeksjoner.
- Kleefstra syndrom (symptomer: tale- og tankeproblemer, muskelsvakhet, søvnforstyrrelser, mangel på oppmerksomhet, åpen munn, hyperaktivitet, anfall, ataksi, balanseforstyrrelser). Kjennetegn: flatt ansikt, kort, stump nese, vidtstående øyne, stor, utadvendt underleppe, aggressive utbrudd.
- Smith-Magenis syndrom (kjennetegnet av anfall, søvnproblemer, intellektuelle og motoriske utviklingsforstyrrelser). Kjennetegn inkluderer et bredt og flatt ansikt og en fremtredende panne.
- Koolen-de Vries syndrom (mild til moderat psykisk utviklingshemming, muskelsvakhet, anfall, vennlighet). Kjennetegn: langt ansikt med høy panne, utstående ører, skjeve øyne, høy leddmobilitet, medfødte hjertefeil.
- Phelan-McDermid syndrom (symptomer: psykisk utviklingshemming, taleforstyrrelser eller mangel på tale). Kjennetegn: store hender med utviklede muskler, muskelsvakhet fra fødselen av, svak svetting.
Slike patologier som adenylsuccinatmangel, autosomalt recessivt mental retardasjonssyndrom 1, kromosom 2q23.1 dupliseringssyndrom, FOXG1, STXBP1 eller MEF2C genhaploinsuffisienssyndromer og noen andre kan "skryte" av symptomer som ligner på Angelman syndrom.
Legens oppgave er å stille en nøyaktig diagnose, skille Angelman syndrom fra patologier med lignende symptomer, og foreskrive effektiv behandling som er relevant for det diagnostiserte stadiet av sykdommen.
Behandling Angelmans syndrom
Angelmans syndrom er en av de patologiene som medisinen fortsatt leter etter effektiv behandling for. Den etiologiske behandlingen av sykdommen er i utviklingsstadiet med ulike metoder og midler, hvorav mange ennå ikke er testet på mennesker. Dette betyr at leger foreløpig må begrense seg til symptomatisk behandling, som på en eller annen måte bidrar til å lindre den lite misunnelsesverdige situasjonen for barn og voksne med marionettsyndrom, som lider av epileptiske anfall, spyttsekresjon, hypotensjon og søvnforstyrrelser.
Dermed er det mulig å redusere hyppigheten og styrken av epileptiske anfall ved hjelp av et riktig valgt antikonvulsivt legemiddel. Men hele vanskeligheten er at anfall hos pasienter med SA skiller seg fra vanlige epileptiske anfall ved at de er preget av flere typer anfall, noe som betyr at tilstanden kan lindres ved å administrere flere legemidler samtidig.
De mest populære antikonvulsiva som brukes til å behandle Angelmans syndrom er: valproinsyre, topiramat, lamotrigin, levetiracetam, klonazepam og legemidler basert på disse. Mindre vanlige er legemidler basert på karmazepin, fenytoin, fenobarbital og etosuksimid, siden noen av dem kan provosere frem en paradoksal effekt som består i å forsterke og øke hyppigheten av epileptiske anfall. Dette skjer hvis legemidlet brukes som en del av monoterapi.
For å behandle sikling brukes vanligvis to metoder: medisinske (legemidler som undertrykker spyttproduksjonen) og kirurgiske metoder, som innebærer reimplantasjon av spyttkanalene. Men i tilfelle av SA anses disse metodene som ineffektive, og spørsmålet forblir åpent. Foreldre og de som tar vare på slike pasienter må være spesielt oppmerksomme på dette problemet, siden pasientene selv vanligvis ikke kontrollerer sikling, og noen rett og slett ikke klarer å ta vare på seg selv.
Et annet problem er kort søvnvarighet. Ofte sover barn med Angelman syndrom ikke mer enn 5 timer, noe som har en negativ innvirkning på hele kroppens funksjon. Lett opphissede, aktive barn som elsker spill og kommunikasjon (selv om de prøver å begrense seg til ikke-verbale metoder) er merkbart slitne i løpet av dagen. For å få en god hvile trenger kroppen en dyp, full søvn, men det er nettopp dette som er haken.
Det ser ut til at beroligende medisiner (fentiaziner og atypiske antipsykotika) som beroliger nervesystemet, burde være tilstrekkelige for å forbedre søvnen hos opphissede pasienter. Men ved AS er bruk av slike medisiner beheftet med risiko for negative effekter. Derfor foretrekker leger fortsatt milde sovepiller, som melatonin (et naturlig hormonelt legemiddel basert på søvnhormonet), som gis til pasienter en time før leggetid i mengden 1 tablett, og difenhydramin. Hyppigheten av administrasjon og dosering bestemmes av legen avhengig av pasientens tilstand og alder.
Noen ganger har pasienter med Angelman syndrom problemer med fordøyelsen og avføringen. Du kan forbedre avføringen med avføringsmidler (helst urtebaserte).
Eller du kan nærme deg problemet annerledes, slik amerikanske leger gjorde, basert på noen metoder for behandling av autisme, fordi mange symptomer som er karakteristiske for AS også er karakteristiske for autisme (impulsivitet, ufrivillige bevegelser, repeterende handlinger, oppmerksomhetsunderskudd, kommunikasjonsproblemer, etc.). Det ble bemerket at introduksjonen av hormonet sekretin, som normaliserer fordøyelsen og avføringen, har en positiv effekt på pasientenes oppmerksomhet, og oksytocin bidrar til å forbedre barnets kognitive evner og hukommelse, og korrekt atferd.
Det er sant at hormoner alene ikke er nok, spesielt ikke når det gjelder barn. Ved Angelman syndrom er atferdsterapi, arbeid med en psykolog og logoped (undervisning i ikke-verbale kommunikasjonsmetoder og tegnspråk) indisert. Opplæringen av slike barn bør baseres på et individuelt program med deltakelse av spesialutdannede lærere, en psykolog og foreldre. Dessverre er ikke dette mulig overalt, og familier blir stående alene med sitt problem.
Siden mange unge pasienter med AS lider av lav muskeltonus og leddproblemer, vies det mye oppmerksomhet til fysioterapi. Oftest tyr leger til bruk av parafinpåføringer, elektroforese og magnetterapi.
Aktiv tonisk massasje og spesielle øvelser innen terapeutisk fysisk trening vil hjelpe det syke barnet til å stå på beina og gå trygt etter en stund. Vanngymnastikk er spesielt nyttig i denne forbindelse, og anbefales for SA i kaldt vann. Det øker muskeltonus og lærer barnet å kontrollere kroppen og koordinere bevegelser.
Antikonvulsiv behandling
Det farligste symptomet på Angelmans syndrom er anfall som ligner på epilepsi. Dette symptomet observeres hos 80 % av pasientene, noe som betyr at alle må foreskrives effektiv antikonvulsiv behandling.
Behandling av epileptiske anfall utføres ved hjelp av vitaminer og antikonvulsiva. Ved Angelman syndrom, ledsaget av konvulsivt syndrom, vil vitaminer fra gruppe B, samt vitamin C, D og E være nyttige. Men å foreskrive vitaminbehandling på egenhånd i dette tilfellet er svært farlig, fordi ukontrollert inntak av vitaminer kan redusere effekten av antiepileptika og provosere frem nye, mer alvorlige og langvarige anfall.
Valg av antikonvulsive legemidler og forskrivning av effektiv dosering bør også gjøres av en spesialistlege. Han eller hun avgjør også om ett legemiddel vil være nok, eller om pasienten må ta to eller flere legemidler over lengre tid.
For de fleste pasienter foreskriver leger valproinsyremedisiner (Valproinsyre, Depakine, Convulex, Valparin, etc.), som forhindrer anfall og forbedrer pasientenes humør og mentale tilstand.
Valproinsyre er tilgjengelig i form av tabletter, sirup og injeksjonsløsninger. Det mest populære legemidlet er depotmedisinen "Depakine" i tabletter og som en løsning for intravenøs administrering. Doseringen av legemidlet bestemmes av legen individuelt avhengig av pasientens vekt, alder og tilstand.
Legemidlet tas under måltider 2 til 3 ganger daglig. Gjennomsnittlig daglig dose er 20–30 mg per kilogram av pasientens vekt, maksimum er 50 mg/kg per dag.
Kontraindikasjoner for bruk. Skal ikke brukes ved lever- og bukspyttkjerteldysfunksjon, hemoragisk diatese, hepatitt, porfyri og overfølsomhet for legemidlet.
Bivirkninger inkluderer håndskjelvinger, fordøyelses- og avføringsforstyrrelser og endringer i kroppsvekt.
«Topiramat» er også et legemiddel for SA. Det produseres i tablettform og brukes både som en del av monoterapi og i kombinasjon med andre legemidler.
Administrasjonsmåte og dosering. Ta tablettene oralt uavhengig av matinntak. Den første daglige dosen for voksne er 25–50 mg, for barn – 0,5–1 mg/kg. Dosen økes hver uke i henhold til legens anvisninger.
Legemidlet bør ikke tas under graviditet og amming, samt ved overfølsomhet for komponentene. Legemidlet har mange forskjellige bivirkninger.
Legemidler som en lege kan foreskrive for Angelman syndrom: Clomazepam, Rivotril, Lamotrigin, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra, etc.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Tradisjonell medisin og homeopati
Tradisjonell medisin, i likhet med homeopatiske preparater, er selvsagt relativt trygg, men effektiviteten av slik behandling for Angelmans syndrom kan anses som kontroversiell.
Selv om folkemedisin fortsatt kan hjelpe med noen ting, snakker vi om å stoppe epileptiske anfall. I denne forbindelse kan urtebehandling være ganske effektiv.
En god effekt gis av en medisinsk samling basert på peon, lakris og andemat (komponentene tas i like mengder). Urtene må males til mel. Etter 2 uker fra starten av inntaket kan du merke en betydelig reduksjon i hyppigheten av anfall.
Lavendelavkok (1 teskje per glass kokende vann) er også nyttig mot kramper. Blandingen kokes i 5 minutter og trekkes i en halvtime. Medisinen tas om kvelden i 14 dager.
En vandig (eller alkoholisk) infusjon av morwort anses som effektiv for epileptiske anfall.
Av de homeopatiske preparatene for å forebygge anfall ved Angelman syndrom kan du bruke medisiner basert på kamille og morwort, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Men det bør tas i betraktning at bare en homeopatisk lege kan foreskrive effektive og trygge doser av legemidler i hvert enkelt tilfelle.
Forebygging
Som leseren sikkert allerede har forstått, er medisinen ennå ikke i stand til å forhindre genmutasjoner og andre kromosomavvik, samt å korrigere situasjonen. Dette kan skje med hvem som helst, fordi barn med Angelmans syndrom er født av friske foreldre, og genetikk, som for tiden er en av de minst studerte grenene av medisin, kan ennå ikke forklare dette.
Det eneste man kan gjøre er å ta en ansvarlig tilnærming til graviditetsplanlegging, registrere seg og gjennomgå undersøkelser i tide. Men igjen, et slikt tiltak vil være mer lærerikt enn forebyggende, som alle undersøkelser. Men unge foreldre vil vite på forhånd hva de skal forberede seg på, og i tilfelle et positivt svar vil de bestemme om de kan ta på seg et slikt ansvar som å oppdra et sykt barn.
Prognose
Prognosen for Angelman syndrom avhenger av kromosomavvikets art og hvor raskt det oppdages. De hardest rammede er de barna hvis kromosom 15 inneholder "hull" i gener (delesjon). Sannsynligheten for at slike pasienter kan gå og snakke er ekstremt lav. Andre tilfeller kan korrigeres med en forsiktig tilnærming og kjærlighet til barnet ditt.
Dessverre vil slike pasienter ikke kunne bli fullverdige medlemmer av samfunnet, til tross for at de langt fra er dumme, de forstår tale og dens betydning. De vil imidlertid ha problemer med kommunikasjon resten av livet. Pasienter kan læres tegnspråk fra barndommen, men de kan ikke tvinges til å kommunisere ved hjelp av ord. Vokabularet til "talende" pasienter er begrenset til minimum av ord som brukes i hverdagen (5-15 ord).
Når det gjelder forventet levealder og generell helsetilstand for pasienter med Angelmans syndrom, svinger tallene her rundt gjennomsnittsverdier. I voksen alder møter pasientene hovedsakelig helseproblemer som skoliose og fedme, som med riktig behandlingstilnærming ikke er livstruende.