Medisinsk ekspert av artikkelen

Nye publikasjoner
Treacher Collins syndrom
Sist anmeldt: 04.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.

Intrauterine forstyrrelser i beinutviklingsprosesser forårsaker alvorlige kraniofasiale deformiteter, og en av variantene av slik patologi er Treacher Collins syndrom (TCS) eller mandibulofascial, det vil si maxillofacial dysostose.
Sykdomskode i henhold til ICD 10: klasse XVII (medfødte anomalier, deformiteter og kromosomale lidelser), Q75.4 - mandibulofacial dysostose.
Fører til Treacher Collins syndrom
Dette syndromet ble oppkalt etter den fremragende britiske øyelegen Edward Treacher Collins, som beskrev hovedtrekkene ved patologien for mer enn hundre år siden. Europeiske leger kaller imidlertid oftere denne typen ansikts- og kjevebensanomali for Franceschettis sykdom eller syndrom – basert på den omfattende forskningen til den sveitsiske øyelegen Adolf Franceschetti, som introduserte begrepet «mandibulofascial dysostose» i midten av forrige århundre. I medisinske kretser brukes også navnet Franceschetti-Collins syndrom.
Treacher Collins syndrom er forårsaket av mutasjoner i TCOF1-genet (ved kromosomlokus 5q31.3-33.3), som koder for et nukleolært fosfoprotein som er ansvarlig for dannelsen av den kraniofaciale delen av det menneskelige embryoet. Som et resultat av en for tidlig reduksjon i mengden av dette proteinet forstyrres biogenesen og funksjonene til rRNA. Ifølge genetikere fra forskningsprogrammet Human Genome fører disse prosessene til en reduksjon i proliferasjonen av embryonale celler i nevralkammen - en ås langs nevralsporet, som lukkes inn i et nevralrør under embryonal utvikling.
Dannelse av ansiktsvev skjer på grunn av transformasjon og differensiering av celler i den øvre (hode) delen av nevralkammen, som migrerer langs nevralrøret til området rundt den første og andre grenbuen i embryoet. Og mangel på disse cellene forårsaker kraniofasiale deformasjoner. Den kritiske perioden for forekomst av anomalier er fra 18 til 28 dager etter befruktning. Ved fullført migrasjon av nevralkammeceller (i den fjerde svangerskapsuken) dannes nesten alt løst mesenkymalt vev i ansiktsområdet, som senere (fra 5 til 8 uker) differensierer til skjelett- og bindevev i alle deler av ansiktet, nakken, strupehodet, øret (inkludert det indre øret) og fremtidige tenner.
Patogenesen
Patogenesen til Treacher Collins syndrom er ofte familiær, og anomalien arves autosomalt dominant, selv om det finnes tilfeller av autosomal recessiv overføring av defekten (med mutasjoner i andre gener, spesielt POLR1C og POLR1D). Det mest uforutsigbare ved kjevedysostose er at mutasjonen arves av barn bare i 40–48 % av tilfellene. Det vil si at hos 52–60 % av pasientene er årsakene til Treacher Collins syndrom ikke assosiert med tilstedeværelsen av en anomali i familien, og det antas at patologien oppstår som et resultat av sporadiske genmutasjoner de novo. Mest sannsynlig er nye mutasjoner konsekvensene av teratogene effekter på fosteret under graviditet.
Blant de teratogene årsakene til dette syndromet nevner eksperter store doser etanol (etylalkohol), stråling, sigarettrøyk, cytomegavirus og toksoplasma, samt glyfosatbaserte herbicider (Roundal, Glyfor, Tornado, etc.). Og listen over iatrogene faktorer inkluderer akne- og seborrheika med 13-cis-retinsyre (Isotretinoin, Accutane); det antikonvulsive legemidlet Fenytoin (Dilantin, Epanutin); psykotrope legemidler Diazepam, Valium, Relanium, Seduxen.
Symptomer Treacher Collins syndrom
De kliniske tegnene på mandibulofascial dysostose og graden av deres uttrykk avhenger i de fleste tilfeller av karakteristikkene ved manifestasjonen av genmutasjoner. Og de første tegnene på denne anomalien er i de fleste tilfeller synlige hos et barn umiddelbart etter fødselen: ansiktet med Treacher Collins syndrom har et karakteristisk utseende. Dessuten er morfologiske anomalier vanligvis bilaterale og symmetriske.
De mest åpenbare symptomene på Treacher Collins syndrom er:
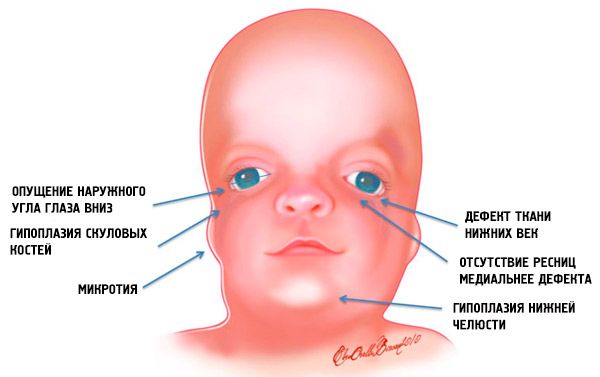
- underutvikling (hypoplasi) av ansiktsbeinene i skallen: zygomatiske, zygomatiske prosesser i pannebenet, laterale pterygoidplater, bihuler, underkjeve og fremspring av beinepifysene (kondyler);
- underutvikling av beinene i underkjeven (mikrognati) og en mandibulær vinkel som er mer stump enn vanlig;
- nesen har normal størrelse, men virker stor på grunn av hypoplasi av superciliarbuene og underutvikling eller fravær av zygomatiske buer i tinningregionen;
- øyespaltene er nedovervendte, det vil si at øynenes form er unormal, med de ytre hjørnene hengende nedover;
- defekter i nedre øyelokk (kolobom) og delvis fravær av øyevipper på dem;
- uregelmessig formede øreskiver med et bredt spekter av avvik, inkludert deres plassering i hjørnet av underkjeven, fravær av fliker, blinde fistler mellom øretragus og munnviken, etc.;
- innsnevring eller lukking (atresi) av den ytre hørselsgangen og anomalier i mellomørets beinknokler;
- fravær eller hypoplasi av parotis spyttkjertlene;
- faryngeal hypoplasi (innsnevring av svelget og luftveiene);
- manglende sammensmelting av den harde ganen (ganespalte), samt fravær, forkortelse eller immobilitet av den myke ganen.
Slike anatomiske avvik har i alle tilfeller komplikasjoner. Dette er funksjonelle hørselsforstyrrelser i form av konduktivt hørselstap eller fullstendig døvhet; synshemming på grunn av feil dannelse av øyeeplene; defekter i ganen forårsaker vanskeligheter med mating og svelging. Det finnes tannokklusjonsforstyrrelser (malokklusjon) forbundet med kjevefeil, som igjen forårsaker problemer med tygging og artikulasjon. Patologier i den bløte ganen forklarer den nasale stemmen.
Komplikasjoner og konsekvenser
Konsekvensene av maxillofaciale anomalier ved Treacher Collins syndrom er at barnets intellektuelle evner ved fødselen er normale, men på grunn av hørselsfeil og andre lidelser observeres sekundær mental retardasjon.
I tillegg føler barn med slike defekter akutt sin underlegenhet og lider, noe som påvirker nervesystemet og psyken deres negativt.
Diagnostikk Treacher Collins syndrom
Postnatal diagnose av Treacher Collins syndrom er i hovedsak basert på kliniske tegn. Kraniofacial dysostose er lett å identifisere når syndromet er fullt uttrykt, men når minimalt uttrykte symptomer på patologi er tilstede, kan det oppstå problemer med å stille riktig diagnose.
I dette tilfellet bør man legge spesiell vekt på vurdering av alle funksjoner forbundet med anomalier, spesielt de som påvirker pusten (på grunn av risikoen for søvnapné). Effektiviteten av mating og hemoglobinoksygenmetning bør også vurderes og overvåkes.
Senere, på den 5.–6. dagen etter fødselen, må omfanget av hørselsskaden bestemmes ved hjelp av audiologisk testing, som bør utføres på fødesykehuset.
En undersøkelse er foreskrevet, hvor instrumentell diagnostikk utføres ved fluoroskopi av kraniofasial dysmorfologi; pantomografi (panoramabilde av beinstrukturen i ansiktsskallen); full kranial computertomografi i forskjellige projeksjoner; CT eller MR av hjernen for å bestemme tilstanden til den indre hørselskanalen.
Den tidligste – prenatale – diagnosen av kjeveavvik ved familiehistorie med Treacher-Collins syndrom er mulig ved biopsi av korionvillus i uke 10–11 av svangerskapet (prosedyren truer med spontanabort og infeksjon i livmoren).
Blodprøver tas også fra familiemedlemmer; ved 16-17 ukers svangerskap analyseres fostervann (transabdominal fostervannsprøve); ved 18-20 ukers svangerskap utføres fosteroskopi og blod tas fra fosterkarene i morkaken.
Men oftest brukes ultralyd i prenatal diagnose av dette syndromet hos fosteret (ved 20-24 ukers svangerskap).
Hvilke tester er nødvendig?
Differensiell diagnose
De samme metodene brukes av spesialister når differensialdiagnostikk er nødvendig for å gjenkjenne det milde Treacher Collins syndromet og skille det fra andre medfødte anomalier i kraniofaciale bein, spesielt: Apert-, Crouzon-, Nager-, Peters-Hewels-, Hellermann-Steph-syndromer, samt hemifaciell mikrosomi (Goldenhar syndrom), hypertelorisme, for tidlig sammenvoksing av kraniale suturer (kraniosynostose) eller nedsatt sammenvoksing av ansiktsbein (kraniosynostose).
Behandling Treacher Collins syndrom
Som i alle tilfeller av genetisk bestemte medfødte defekter, er behandlingen av alvorlige former for Treacher Collins syndrom utelukkende palliativ, siden det rett og slett ikke finnes terapeutiske metoder for slike patologier. Spekteret og graden av deformasjoner i dette syndromet er omfattende, og derfor har arten og intensiteten av medisinsk intervensjon også mange alternativer.
Høreapparater brukes til å korrigere og forbedre hørselen, og logopeditimer brukes til å forbedre talen.
Kirurgiske inngrep er nødvendige i tidlig alder ved alvorlige tilfeller av innsnevring av luftveiene (trakeostomi utføres) og strupehode (gastrostomi utføres for mating). Kirurgisk korreksjon av ganen kan også være nødvendig.
Mandibulær forlengelsesoperasjoner utføres i alderen 2–3 år eller senere. Bløtvevsrekonstruksjon inkluderer korrigering av kolobom i nedre øyelokk og plastisk kirurgi på øret.
Forebygging
Prognose
Hva er prognosen for denne patologien? Det avhenger av graden av deformasjon og intensiteten av symptomene. Treacher Collins syndrom er en livslang diagnose.
 [ 25 ]
[ 25 ]