Medisinsk ekspert av artikkelen

Nye publikasjoner
Pierre Robin-syndromet
Sist anmeldt: 04.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.

Pierre Robins syndrom, også kjent i medisin som Robin-anomali, er en medfødt patologi i utviklingen av kjevedelen av ansiktet. Sykdommen fikk navnet sitt til ære for den franske tannlegen P. Robin, som først beskrev alle tegnene. Lannelongue og Menard beskrev først Pierre Robins syndrom i 1891 i sin rapport om to pasienter med mikrognati, ganespalte og retroglossoptose. I 1926 publiserte Pierre-Robin et tilfelle av sykdommen hos et spedbarn med tegn på det klassiske syndromet. Frem til 1974 var triaden av tegn kjent som Robin-Pierre syndrom. Imidlertid brukes dette syndromet nå til å beskrive misdannelser med samtidig tilstedeværelse av flere anomalier.
Epidemiologi
Det er en heterogen medfødt defekt som har en prevalens på 1 av 8500 levendefødte. Forholdet mellom menn og kvinner er 1:1, bortsett fra den X-bundne formen.
Blant disse pasientene har 50 % av spedbarn en ufullstendig bløt ganespalte, resten er født med en buet og uvanlig høy ganespalte, men uten ganespalte.
Fører til Pierre Robin-syndromet
Muligheten for autosomal recessiv arv av sykdommen vurderes. Det finnes to typer syndrom avhengig av etiologien: isolert og genetisk bestemt. Den isolerte typen utvikler seg på grunn av kompresjon av den nedre delen av kjeven under embryonal utvikling. Kompresjon kan utvikle seg på grunn av:
- Tilstedeværelsen av lokaliserte seler i livmoren (cyster, arr, svulster).
- Flerfoldsgraviditet.
I tillegg kan utviklingen av kjeven hos fosteret forstyrres av:
- Virusinfeksjoner som den vordende moren led av under graviditeten.
- Nevrotrofiske lidelser.
- Utilstrekkelig mengde folsyre i kroppen til en gravid kvinne.
Patogenesen
Pierre Robins syndrom er forårsaket av embryonale lidelser som er forårsaket av et bredt spekter av patologier i prenatalperioden.
Det finnes tre patofysiologiske teorier som kan forklare forekomsten av Pierre Robins syndrom.
Mekanisk teori: Denne teorien er den mest sannsynlige. Underutvikling av mandibulærapparatet forekommer mellom 7. og 11. svangerskapsuke. Tungens høye plassering i munnhulen fører til dannelse av ganespalter, som gjør at hulvenen ikke lukkes. Denne teorien forklarer den klassiske omvendte U-formede kløften og fraværet av den tilhørende leppespalten. Oligohydramnion kan spille en rolle i etiologien, siden fravær av fostervann kan føre til deformasjon av haken og påfølgende kompresjon av tungen mellom hulvenene.
Nevrologisk teori: Forsinkelse i nevrologisk utvikling er observert med elektromyografi av musklene i drøvelen og faryngeusøylen, og smak på grunn av ledningsforsinkelse i nervus hypoglossus.
Teori om dysnevroregulering av rombensephalon: Denne teorien er basert på forstyrrelsen av utviklingen av rombensephalon under ontogenesen.
Utilstrekkelig utvikling av den nedre delen av barnets kjeve fører til at munnhulen reduseres betydelig. Dette forårsaker igjen den såkalte pseudomakroglossia, det vil si at tungen forskyves til baksiden av svelgveggen. Denne patologien fører til utvikling av luftveisobstruksjon.
Så lenge babyen gråter eller beveger seg, forblir luftveiene frie, men så snart babyen sovner, oppstår obstruksjonen igjen.
På grunn av luftveisforstyrrelser er prosessen med å mate babyen svært vanskelig. På dette tidspunktet oppstår nesten alltid luftveisobstruksjon. Hvis ingen medisinsk korreksjon utføres, kan en slik patologi føre til alvorlig utmattelse av hele kroppen og til og med død.
Symptomer Pierre Robin-syndromet
Sykdommen er preget av tre hovedsymptomer:
- Nedre mikrognati (underutvikling av underkjeven, forekommer i 91,7 % av tilfellene av sykdommen). Den kjennetegnes av at den nedre tannbuen trekkes tilbake 10–12 mm bak den øvre tannbuen. Underkjeven har en liten kropp, en stump vinkel. Barnet oppnår normal utvikling ved omtrent 5–6 års alder.
- Glossoptose (tilbaketrekking av tungen på grunn av utilstrekkelig utvikling, observert i 70-85 % av tilfellene).
- Makroglossi og ankyloglossi er relativt sjeldne symptomer, observert i 10–15 % av tilfellene.
- En sprekk dukker opp på himmelen.
- Bradypné og dyspné.
- Mild cyanose.
- Kvelning, som oftest oppstår under forsøk på å mate babyen.
- Å svelge er umulig eller svært vanskelig.
- Føler meg som å kaste opp.
- Aurikulære anomalier i 75 % av tilfellene.
- Konduktivt hørselstap forekommer hos 60 % av pasientene, mens atresi i den ytre hørselsgangen forekommer hos bare 5 % av pasientene, utilstrekkelig pneumatisering av mastoidhulen i temporalbenet.
- Anomalier i det indre øret (aplasi i de laterale halvsirkelformede kanalene, den store vestibulære akvedukten, tap av hårceller i cochlea).
- Nesemisdannelser er uvanlige og består hovedsakelig av anomalier i neseroten.
- Tannmisdannelser forekommer i 30 % av tilfellene. Laryngomalasi og velofaryngeal insuffisiens forekommer hos omtrent 10–15 % av pasienter med Pierre Robins syndrom.
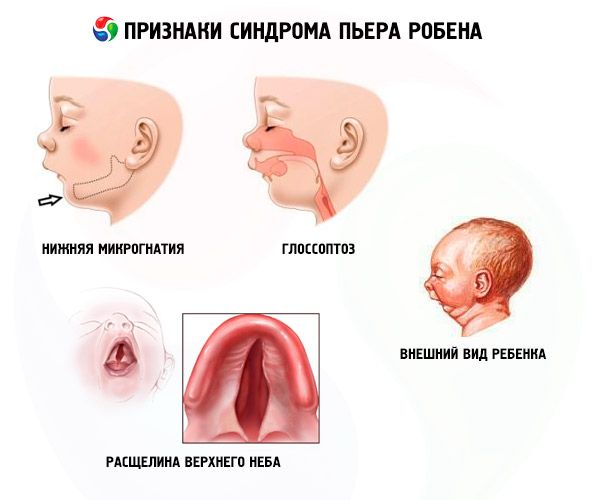
Systemiske trekk ved Pierre Robins syndrom
Systemiske utviklingsanomalier er beskrevet i 10–85 % av registrerte tilfeller.
Øyeavvik forekommer hos 10–30 % av pasientene. Disse kan omfatte: hypermetropi, nærsynthet, astigmatisme, hornhinnesklerose og stenose av kanalen nasolakrimal.
Kardiovaskulære patologier: godartede hjertebilyd, lungearteriestenose, åpen ductus arteriosus, ovalt vindu, atrieseptumdefekt og pulmonal hypertensjon. Forekomsten varierer fra 5–58 %.
Anomalier relatert til muskel- og skjelettsystemet (70–80 % av tilfellene): syndaktyli, dysplastiske falanger, polydaktyli, klinodaktyli, hypermobilitet i ledd og oligodaktyli i øvre lemmer. Anomalier i nedre lemmer: fotanomalier (klumpfot, metatarsal adduksjon), femurmisdannelser (valgus- eller varusbekken, korte femurer), hofteanomalier (medfødt dislokasjon, kontrakturer), kneleddsanomalier (GENU VALGUS, synkondrose). Misdannelser i ryggsøylen: skoliose, kyfose, lordose, vertebral dysplasi, agenesi av korsbenet og sinus coccygeal.
Patologi i sentralnervesystemet: epilepsi, forsinkelser i nervesystemets utvikling, hydrocephalus. Hyppigheten av CNS-defekter er omtrent 50 %.
Urinveiene: ikke-nedsokte testikler (25 %), hydronefrose (15 %) og hydrocele (10 %).
Tilknyttede syndromer og tilstander: Sticklers syndrom, trisomi 11q-syndrom, trisomi 18, 4q-delesjonssyndrom, revmatoid artropati, hypokondroplasi, Möbius syndrom.
Stages
Det er tre stadier av sykdommens alvorlighetsgrad, som avhenger av tilstanden til barnets luftveier:
- Mild - det er mindre problemer med mating, men pusten er nesten uproblematisk. Behandlingen utføres poliklinisk.
- Moderat – pusten er moderat vanskelig, det er moderat vanskelig å mate barnet. Behandlingen utføres på sykehus.
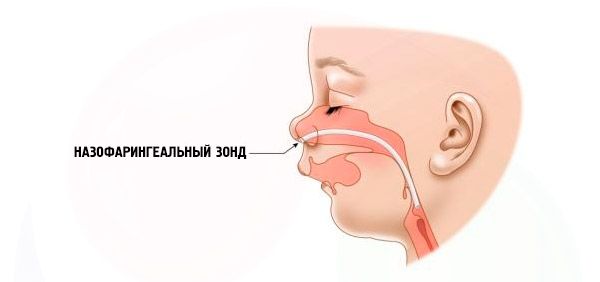
- Alvorlig – pusten er svært vanskelig, barnet kan ikke mates normalt. Det er nødvendig å bruke spesielle enheter (intranasal sonde).
Komplikasjoner og konsekvenser
Kombinasjonen av mikrognati og glossoptose kan føre til alvorlige respiratoriske komplikasjoner og problemer under mating av barnet.
Pierre Robins syndrom forårsaker følgende komplikasjoner:
- Stridosepust på grunn av luftveisobstruksjon. Laryngomalasi eller søvnasfyksi.
- Barnets psykomotoriske utvikling henger langt etter jevnaldrende.
- Den fysiske utviklingen henger også etter.
- Pasientenes tale er svekket.
- Hyppige ørebetennelser som blir kroniske og fører til hørselshemming.
- Obstruktiv søvnapnésyndrom, forekomsten av død i søvn varierer i 14–91 % av tilfellene.
- Problemer med tenner.
Diagnostikk Pierre Robin-syndromet
Diagnosen av Pierre Robins syndrom er ikke vanskelig. Den er basert på kliniske manifestasjoner. For å utelukke andre patologier er det svært viktig å konsultere en genetiker.
Barn med Robins medfødte misdannelse har pusteproblemer fra fødselen av på grunn av at tungen stadig synker tilbake. Babyen er urolig, huden er blåaktig, og det kommer piping fra brystet ved innånding. Kvelning kan forekomme under mating. Diagnosen kan også stilles ved barnets uvanlige utseende - "fugleansikt". Ofte utvikler pasienter andre defekter: nærsynthet, grå stær, patologi i kjønnsorganene, hjertesykdom, anomalier i ryggradens utvikling.
Basert på disse kliniske manifestasjonene vil det ikke være vanskelig for en spesialist å stille en korrekt diagnose.
Hvem skal kontakte?
Behandling Pierre Robin-syndromet
Behandlingen utføres umiddelbart etter fødselen av et barn med Pierre Robin syndrom. Hvis sykdommen er mild, er det nødvendig å holde barnet vertikalt eller liggende på magen for å forbedre pasientens tilstand. Barnets hode bør vippes mot brystet. Under mating anbefales det ikke å holde barnet i horisontal stilling slik at mat ikke kommer inn i luftveiene.
Hvis underutviklingen av underkjeven er ganske uttalt, brukes kirurgisk inngrep for å bringe den tilbaketrukne tungen til en normal fysiologisk posisjon. I alvorlige tilfeller trekkes tungen opp og festes på underleppen. I svært alvorlige tilfeller må trakeostomi, glossopeksi og distraksjonsosteogenese av underkjeven utføres.
Konservativ behandling brukes også.
Medisiner
Fenobarbital. En sovepille og beroligende medisin, har en antikonvulsiv effekt. Hver tablett inneholder 100 ml fenobarbital. Doseringen er individuell, da den avhenger av alvorlighetsgraden av sykdommen og barnets tilstand. Legemidlet er forbudt for pasienter med leversvikt, hyperkinesi, anemi, myasteni, porfyri, diabetes mellitus, depresjon og intoleranse mot komponentene. Følgende symptomer er mulige ved bruk: svimmelhet, asteni, hallusinasjoner, agranulocytose, kvalme, lavt blodtrykk og allergier.
Klonazepam. Et legemiddel foreskrevet for behandling av epilepsi. Legemidlet inneholder virkestoffet klonazepam, som er et benzodiazepinderivat. Det har antikonvulsive, angstdempende og muskelavslappende effekter. Dosen bestemmes av behandlende lege, men bør ikke overskride maksimumsdosen - 250 mcg per dag. Skal ikke tas ved søvnløshet, muskelhypertoni, psykomotorisk agitasjon, panikklidelser. Følgende symptomer er mulige ved bruk: sløvhet, kvalme, dysmenoré, hodepine, leukopeni, urinretensjon eller inkontinens, hårtap, allergi.
Sibazon. Tilgjengelig i form av en løsning og rektaltabletter. Det aktive stoffet er et benzodiazepinderivat (sibazon). Det har en beroligende, angstdempende og antikonvulsiv effekt. Doseringen er individuell. Pasienter med kronisk hyperkapni, myasteni og benzodiazepinintoleranse er forbudt å ta legemidlet. Ved bruk av legemidlet kan følgende symptomer utvikles: kvalme, forstoppelse, hodepine, svimmelhet, hikke, urininkontinens og allergier.
Cortexin lyofilisat. Et legemiddel med nootropisk effekt. Legemidlet inneholder et kompleks av vannløselige polypeptidfraksjoner og glysin. Doseringen er individuell og foreskrives av behandlende lege i samsvar med pasientens tilstand. Pasienter med intoleranse mot cortexin er forbudt å ta legemidlet. Legemidlet kan forårsake allergiske reaksjoner.
Fysioterapibehandling
Vanligvis brukes posisjonsterapi i milde stadier av syndromet, der barnet plasseres på magen i oppreist stilling inntil tyngdekraften tvinger underkjeven til å vokse riktig.
Kirurgisk behandling
Kirurgisk behandling brukes primært til å korrigere glossoptose. Det finnes flere metoder:
- Støtte av tungen med en sølvtråd. Tråden føres gjennom den nedre delen av tannkjøttet og underleppen. Metoden kalles Douglas.
- Duhamels metode – en tykk sølvtråd føres gjennom pasientens tungebase og begge kinn. Skal ikke brukes lenger enn tretti dager.
- Ortopediske apparater for tungeforlengelse og fiksering.
- Ved ett års alder kan man operere for å korrigere ganespalte.
Prognose
Prognosen og sykdomsforløpet er alvorlig. Oftest inntreffer døden i løpet av de første levedagene i moderat og alvorlig stadium av sykdommen (årsaken er kvelning). Risikoen for død i det første året er også ganske høy på grunn av en rekke infeksjoner.
For pasienter over to år er prognosen gunstig.
 [ 36 ]
[ 36 ]