Medisinsk ekspert av artikkelen

Nye publikasjoner
Araknodaktylitet
Sist anmeldt: 04.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
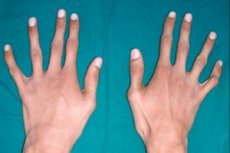
En av de sjeldne arvelige bindevevspatologiene er araknodaktyli – en deformasjon av fingrene, ledsaget av forlengelse av rørformede bein, skjelettkrumninger, forstyrrelser i det kardiovaskulære systemet og synsorganene.
Sykdommen ble først beskrevet av Dr. Williams og den franske barnelegen Marfan, som kalte den dolichostenomeli. Senere dukket begrepet Marfan syndrom opp i verdensmedisinen, og siden 1902 har navnet araknodaktyli blitt brukt. Patologien er relatert til medfødte bindevevsforstyrrelser, og et av de viktigste tegnene anses å være forlengede, tynne og buede "edderkopp"-fingre. [ 1 ]
Epidemiologi
Araknodaktyli regnes som en arvelig monogen patologi av bindevevet, med autosomal dominant arv, høy penetrans og varierende grad av ekspressivitet.
I omtrent 75–80 % av tilfellene er sykdommen arvelig, og i de resterende tilfellene oppstår den som et resultat av spontane mutasjoner (spesielt på grunn av punkt-missense-mutasjoner). [ 2 ]
Det er bevist at de patogenetiske trekkene ved araknodaktyli er forårsaket av endringer i flere gener (i 95 % av tilfellene):
- i fibrillin-1-genet (omtrent tusen mutasjoner er studert) på kromosom 15q21.1;
- TGFβR1- eller TGFβR2-genet på kromosom 9 og 3p24.2-P25.
Disse endringene påvirker direkte omfanget av kliniske manifestasjoner.
Mutasjoner i α-2-kjeden av type I kollagen er beskrevet hos 5 % av pasientene.
Forekomsten av araknodaktyli er omtrent 2 tilfeller per 5000 personer. Det finnes ingen rasemessig eller kjønnsmessig bestemmelse. [ 3 ]
Fører til araknodaktylitet
Araknodaktyli oppstår som et resultat av spontan mutasjon. Sykdommen er ikke karakterisert av de allment kjente tegnene på medfødt arvelig patologi, og forlengede fingre manifesterer seg hovedsakelig ved Marfan syndrom og homocystinuri.
Sykdommen er sjelden, og alle pasientene som ble funnet hadde en mutasjon av FBN1-genet (fibrillin-genet) lokalisert på kromosom 15 i q21.1-regionen. Den kliniske polymorfismen til syndromet kan forklares med mengden mutasjoner, hvor omtrent 15 % er mutasjonsendringer som oppsto under unnfangelsen. [ 4 ]
Atypiske varianter av araknodaktyli er forårsaket av mutasjoner i andre gener: det er oppdaget en sammenheng mellom utviklingen av patologien og mutasjoner i LTBP3, R-genet, som er lokalisert på kromosom 14 i q24-regionen og transformerer vekstfaktor.
Det overveldende flertallet av mutasjoner er såkalte missense-mutasjoner (omtrent 57 %). Mer enn 20 % er små delesjoner, med tap av kromosomale deler, omtrent 12 % er spleisingsstedsmutasjoner, 8 % er nonsensmutasjoner, og 2 % er storskala omorganiseringer og delesjoner. [ 5 ]
Medfødt kontraktural araknodaktyli er forårsaket av en forstyrrelse i syntesen av proteinagensen fibrillin-2. Problemet er forårsaket av en mutasjon i FBN2-genet, som ligger på det femte kromosomet i q23-q31-regionen. Alle mutasjoner er hovedsakelig punktmutasjoner, som bytter kodonet til å kode for en annen aminosyre. [ 6 ]
Homocystinuri utvikler seg mot bakgrunn av nedsatt metabolisme av den essensielle aminosyren metionin. Årsaken til patologien kan være tap av aktivitet eller reduksjon av følgende enzymstoffer:
- Enzym cystathionin beta-syntase (CBS). Patologien er B6 resistent og manifesterer seg med tegn som er karakteristiske for den B6 uavhengige formen, men introduksjonen av et B6 vitaminpreparat fører ikke til bedring.
- Enzym N5, N10 metylentetrahydrofolatreduktase (MTHFR). Dette enzymatiske stoffet er et substrat som forårsaker demetylering av homocystein og dets omdannelse til metionin, en essensiell aminosyre som er en komponent i mange protein- og peptidstoffer.
- Enzymet N5 metylentetrahydrofolat. Vi snakker om en B6 avhengig form for patologi, som utvikler seg ved mangel på vitamin B6 . Innføring av vitaminpreparat i kroppen gjør det mulig å forbedre pasientens tilstand.
- Homocystein-transmetylase-enzym. Utvikles mot bakgrunn av metabolske forstyrrelser i kobalamin – vitamin B12.
Risikofaktorer
Araknodaktyli er en genetisk betinget sykdom karakterisert ved systemisk skade på bindevevet. Patologien er assosiert med endringer i fibrillin 1-genet, lokalisert på den korte armen av kromosom 15 i lokus 21.1.
Araknodaktyli arves autosomalt dominant, med høy penetrans og variabel ekspressivitet. Patologien kan manifestere seg både på mannlig og kvinnelig side.
Patogenesen
Mer enn 50 % av menneskekroppens masse består av bindevevsstrukturer, spesielt skjelett, hud, karnettverk, lymfe og blod.
Bindevevsceller er representert av fibroblaster og deres undertyper: osteoblaster, kondrocytter, keratoblaster, odontoblaster, samt makrofager og mastceller.
Embryonalt bindevev er materialet for dannelsen av konstitusjonelle, genetiske og epigenetiske komponenter. Bindevevssykdommer gjenspeiles i en eller annen grad på hele kroppen som helhet, på dens funksjonelle kapasitet og konstitusjon.
Ved araknodaktyli skjer det en substitusjon av nukleotider i genet som inneholder strukturell informasjon om fibrillin-1-peptidet. Dette proteinstoffet tilhører glykoproteingruppen, deltar i det mikrofibrillære systemet og danner grunnlaget for elastiske fibriller i bindevevet.
Vevet opprettholder en stabil struktur på grunn av den intercellulære matrisen, som inneholder mange vekstfaktorer som sikrer systematisk cellefornyelse. Store kar og leddbånd inneholder en rekke elastinfibriller, hvis skade forårsaker de viktigste symptomene på araknodaktyli.
Den transformerende vekstfaktoren β er betydelig skadet: dens inaktive form er ikke bundet, noe som fører til en økning i den biologiske aktiviteten til denne faktoren.
Fibrillære lidelser fører til unormal dannelse av fibre, noe som resulterer i tap av styrke og elastisitet i huden og andre bindevevsstrukturer.
Når kollagenstrukturen forstyrres, forstyrres den primære lenken i hemostasen.
Eksperter bemerker at hormonelle ubalanser er en del av utviklingen og utviklingen av anomalier i bindevevsstrukturer, inkludert araknodaktyli. Trombotiske symptomer bestemmes av reologiske lidelser - spesielt endringer i blodets viskositet i de modifiserte karene i brachiocephalic-regionen.
Fordøyelseskanalen påvirkes på grunn av kollagenforstyrrelser: hypomotorisk biliær dyskinesi, brokk i spiserørets åpning av mellomgulvet, hepatobiliære anomalier, kronisk form for "slettet" gastroduodenitt, dolichosigma observeres.
Hvordan arves araknodaktyli?
Araknodaktyli arves som en autosomal dominant egenskap, men kan også oppstå som et resultat av spontane mutasjoner. Det vil si at patologien ikke nødvendigvis trenger å manifestere seg hos en av de nære slektningene – for eksempel hos foreldre, besteforeldre. Mutasjoner som ligger til grunn for patologien kan gjøre seg kjent tilfeldig.
Symptomer araknodaktylitet
Innen medisin skilles det mellom latent og åpenbar araknodaktyli. Ved latent araknodaktyli er det forandringer, men de er isolerte og begrenset til ett eller to organsystemer. Åpenbar araknodaktyli manifesterer seg i flere synlige lidelser med varierende intensitet av symptomene. I dette tilfellet kan pasientens tilstand være relativt stabil i flere tiår, eller utvikle seg jevnt og trutt, og manifestere seg i forhold til andre organer og systemer.
Ofte kan arachnodactyly syndrom oppdages i løpet av de første dagene etter babyens fødsel.
Det viktigste ytre tegnet på araknodaktyli er tynne og langstrakte fingre, som på grunn av korte sener får en typisk krumning og ligner edderkoppben.
Edderkoppfingre eller araknodaktyli blir tydelig synlige nesten fra nyfødtperioden. Men tegnet blir tydeligere rundt treårsalderen.
Vekstindikatorer er også karakteristiske: barn er vanligvis høye, underarmer og leggbein er lange, lemmene er uforholdsmessige og tynne. Uforholdet er spesielt tydelig ved den høye lokaliseringen av midjen og kneskålene. [ 7 ]
Andre skjelettlidelser inkluderer:
- innsnevret og langstrakt hodeskalleform (dolichocephalic) med et unormalt formet ansiktsområde;
- traktformet konfigurasjon av brystet (den såkalte "fuglen");
- buet ryggrad;
- medfødte hofteluksasjoner;
- vanlige kne-skulder-dislokasjoner;
- krumning av hælbenet;
- osteofyttformasjoner – beinvekster;
- flate føtter;
- utilstrekkelig utvikling av fettlaget.
Ytterligere tegn på araknodaktyli kan omfatte:
- nærsynthet;
- blåaktig senehinne;
- subluksasjon av linsen;
- hjerte- og blodåresykdommer (hjertefeil, aortaaneurisme);
- uttalt tynnhet;
- hypermobilitet i ledd;
- "buet" himmel.
Araknodaktyli kjennetegnes av forlengede rørformede bein, noe som forårsaker ulike skjelettkrumninger. I tillegg har mange pasienter pyramideformede lidelser, asymmetri i senereflekser, anosokoria og nystagmus på bakgrunn av normal mental utvikling.
Hvis vi snakker om araknodaktyli, ledsagende homocystinuri, kan pasienten oppleve følgende symptomer:
- periodiske anfall av kramper;
- sekundær glaukom med linsesubluksasjon;
- netthinneavløsning, astigmatisme;
- skade på arterielle stammer (inkludert nyrer, hjerne);
- osteoporose;
- trombose;
- psykisk utviklingshemming.
Araknodaktyli hos nyfødte
I de fleste tilfeller kan araknodaktyli identifiseres hos et barn i løpet av de første dagene av livet. Men med alderen har symptomene en tendens til å forverres.
De første tegnene som gir mistanke om sykdommen er vanligvis følgende:
- uvanlig lange lemmer og fingre;
- undervektig i tilstrekkelig fysisk form hos barnet;
- langstrakt, langt ansikt;
- mangel på kroppsfett, dårlig utviklede muskler;
- overdreven fleksibilitet i leddene.
Fra omtrent fire års alder, mot bakgrunnen av araknodaktyli, begynner brystet å forandre seg, ryggsøylen blir buet, og det utvikles flate føtter.
Når det gjelder synsorganene, observeres nærsynthet, linseekstopi, endret hornhinnekonfigurasjon, strabismus, hypoplastiske prosesser i iris og netthinne. Slike lidelser kan oppdages allerede i de første 2-3 årene av et barns liv, selv om de utvikler seg over årene.
Den største faren utgjør forstyrrelser i det kardiovaskulære systemet. Hvis slike lidelser er tilstede, er det høy risiko for at pasienten dør allerede i barndommen i mangel av passende behandling. Blant de spesielt farlige patologiene kan man skille ut skader på karveggene, hjertemisdannelser og defekter i koronarkarene. Noen ganger, hos barn i det første leveåret, oppdages økende hjertesvikt allerede.
Araknodaktyli kan være ledsaget av skade på andre organer, spesielt nervesystemet, bronkiene og lungene, huden og det urogenitale systemet. Slike lidelser oppdages under diagnostiske prosedyrer.
Komplikasjoner og konsekvenser
De vanligste bivirkningene av araknodaktyli er skade på hjertet og luftveiene. I tillegg oppstår problemer med synet, helt opp til og med tap av synet.
Pasienter med araknodaktyli krever kompleks behandling av alle eksisterende patologier. Hvis slik behandling ikke utføres, reduseres pasientenes forventede levealder betydelig: sjeldne pasienter klarer å leve til 40 år uten medisinsk inngrep. Over årene forverres problemene med det kardiovaskulære systemet, og risikoen for omfattende blødninger med tragisk utfall øker.
Personer som lider av araknodaktyli bør gjennomgå regelmessige undersøkelser og behandle samtidige patologier: bare under denne tilstanden har de all sjanse til å leve et langt liv uten alvorlige komplikasjoner.
Marfans syndrom og araknodaktyli krever spesiell oppmerksomhet dersom den kvinnelige pasienten blir gravid. I en slik situasjon er det viktig å undersøke pasienten regelmessig og grundig, og å gi medisinsk støtte til tilstanden til hennes hjerte- og karsystem. Dette er den eneste måten å garantere et vellykket svangerskap og en trygg fødsel av barnet.
For å unngå bivirkninger bør pasienter med araknodaktyli undersøkes regelmessig av leger for å forhindre utvikling av kardiovaskulære og muskuloskeletale patologier. De anbefales å leve en utelukkende sunn livsstil med moderat fysisk aktivitet.
Diagnostikk araknodaktylitet
Klinisk diagnostikk for araknodaktyli inkluderer innsamling av plager, familiehistorie, fenotypevurdering, samt antropometriske og fysiske undersøkelser. Det er viktig å bestemme tilstedeværelsen av manifestasjoner av bindevevspatologier hos direkte slektninger: for dette formålet utføres en genealogisk studie. [ 8 ]
Laboratoriediagnostikk for araknodaktyli innebærer å studere tilstanden til visse typer bindevev, representert av bein og bruskvev, blod og lymfe. Den mest informative anses å være en studie av nivåene av oksyprolin og glykosaminoglykaner i det daglige urinvolumet. I tillegg vurderes innholdet av lysin, prolin og oksyprolin i blodet. For å bestemme endrede forhold mellom forskjellige typer kollagener og strukturelle abnormiteter i kollagenfibre, foreskrives typing ved hjelp av metoden med indirekte immunofluorescens ved bruk av polyklonale antistoffer mot kollagen og fibronektin. [ 9 ] Avhengig av indikasjonene utføres følgende tester for å gjenspeile kvaliteten på metabolske prosesser i bindevevet:
- glykosaminoglykaner, fibrillin, fibronektin;
- studie av hydroksyprolin, markører for type 1 kollagenbiosyntese, aminoterminale propeptider av type 1 prokollagen, markører for type 1 kollagennedbrytning, galaktosyloksylysin, deoksypyridinolin;
- analyse av regulering av kollagenmetabolisme (studie av matriksmetalloproteinaser og vevshemmere av matriksmetalloproteinaser, transformerende vekstfaktor);
- analyse av mikro- og makroelementer (innhold av kalsium og magnesium, fosfor, jern, svovel og kobber, kobolt og selen, sink og mangan, fluor og vanadium, silisium og bor);
- studie av markører for beinvevsdannelse og hastigheten på ombyggingsprosesser (analyse av innholdet av osteocalcin, alkalisk fosfatase i bein, parathyroidhormon, somatotropisk hormon, prolaktin, vitamin D3 , pentosidin, homocystein i urin og blod).
Instrumentell diagnostikk har som mål å identifisere eksisterende utviklingsdefekter og vurdere den funksjonelle tilstanden til organer og systemer. Kardiovaskulære lidelser ved araknodaktyli krever obligatorisk inkludering i diagnostikklisten av slike prosedyrer som:
- elektrokardiografi;
- 24-timers EKG-overvåking;
- ultralydundersøkelse av hjertet og blodårene.
Vaskulære anomalier undersøkes ved hjelp av computertomografi eller magnetisk resonansavbildning. Ytterligere diagnostiske prosedyrer kan omfatte:
- Ultralyd av mageorganer, urogenitalsystemet;
- Røntgen av brystorganene, hofteleddene; [ 10 ]
- CT eller MR av ryggraden.
Etter alle nødvendige tester henvises pasienten til genetisk veiledning.
Differensiell diagnose
Det er en fenotypisk likhet mellom Beals syndrom og araknodaktyli ved Marfan syndrom (mutasjoner av henholdsvis FBN2- og FBN1-genene), som skyldes den nesten fullstendige identiteten til proteinstoffene fibrillin 1 og fibrillin 2. Det er ikke overraskende at det andre navnet på Beals syndrom er kontraktur-araknodaktyli. [ 11 ]
Differensiering utføres også med andre bindevevspatologier:
- Sticklers syndrom;
- Ehlers-Danlos syndrom;
- homocystinuri;
- artrogrypose.
Beals syndrom er karakterisert av en marfanoid fenotype, medfødte fleksjonskontrakturer i store og små ledd, araknodaktyli i håndledd og fot, krumning av ryggraden og unormal øreform (såkalte «krøllede» ørestokker). Molekylær analyse av FBN2-genet utføres for å bekrefte diagnosen.
Artrogrypose er karakterisert av flere leddkontrakturer, kort vekst og redusert intelligens. Formen på fingrene og ørene er uanselig. [ 12 ]
Ehlers-Danlos syndrom er karakterisert av kyfoskolose, traktformet krumning av brystet, uttalt platføtter og nedsatt refraksjon. Økt mobilitet og leddforskyvninger, høy hudutvidelse og følsomhet er typiske.
Sticklers syndrom er preget av endringer i leddvolum og stivhet.
Hvem skal kontakte?
Behandling araknodaktylitet
Araknodaktyli er en genetisk patologi, og medisinen har for tiden ingen metoder for å korrigere genfeil. Derfor er behandlingen rettet mot å optimalisere pasientens tilstand, forhindre forverring av patologien og eliminere symptomatiske manifestasjoner. Kompleks terapi er foreskrevet, som involverer flere medisinske spesialister samtidig, avhengig av typen av de mest uttalte symptomene: ofte er det nødvendig å i tillegg kontakte en ortoped, kardiolog, øyelege, gastroenterolog og leger med andre spesialiteter. [ 13 ]
Blant anbefalingene for klinisk behandling av pasienter anses følgende som generelle prinsipper:
- begrense fysisk aktivitet, men ikke eliminere den fullstendig (som en del av å støtte det kardiovaskulære systemet);
- medikamentell behandling;
- om nødvendig – kirurgisk korreksjon av de mest berørte områdene av hjertet og blodårene;
- ortopedisk korreksjon;
- spabehandling, fysioterapi, terapeutisk trening.
Kostholdet til pasienter med araknodaktyli bør inneholde en tilstrekkelig mengde proteinrike produkter beriket med mikronæringsstoffer, vitaminer og fettsyrer. Det anbefales å spise kjøtt, fisk, sjømat, bønner og nøtter. Hvis araknodaktyli er forårsaket av homocystinuri, er inntaket av animalsk protein sterkt begrenset.
Barn med tynn kroppsbygning og høy vekst anbefales å introdusere bomullsfrøolje og soyaolje, solsikkefrø, svinefett og smult i kostholdet sitt fra tidlig alder. I tillegg tilbys preparater som inneholder flerumettede fettsyrer som omega, som hemmer produksjonen av somatotropisk hormon.
For å normalisere proteinmetabolismen foreskrives vitaminer fra gruppen B. De kan også fås fra matvarer: bokhvete, erter, lever.
Det er svært viktig at pasienten får askorbinsyre i kroppen sammen med maten. For dette formålet er nypeinfusjon, paprika, kål, sitrusfrukter, samt havtorn og purre nødvendigvis inkludert i kostholdet.
Om nødvendig tilbys pasienter ortopedisk korreksjon, som er nødvendig for å redusere belastningen på ryggraden og leddene. Til dette formålet brukes ortopediske sko, kne- og andre hjelpemidler, innleggssåler og elastiske bandasjer.
Kirurgisk behandling utføres i henhold til indikasjoner.
Medisiner
Medikamentell behandling for araknodaktyli utføres 1–2 ganger i året, avhengig av pasientens tilstand og alvorlighetsgraden av patologiske symptomer. Behandlingsvarigheten bestemmes individuelt og er i gjennomsnitt 4 måneder. [ 14 ]
For å stimulere kollagendannelse foreskrives følgende legemidler: Piaskledin 300, L-lysin eller L-prolin i kombinasjon med komplekse multivitaminpreparater som inneholder askorbinsyre, B-vitaminer, tokoferol, magnesium, sink, selen, kobber.
Blant kondroprotektorer er den mest optimale bruken kondroitinsulfat og glukosaminsulfat - legemidler som deltar i reguleringen av kondrocyttmetabolisme, undertrykkelse av enzymsyntese, økning i kondrocytters følsomhet for enzymatisk virkning, stimulering av anabilistiske prosesser, etc. Kombinerte midler anses som optimale slike legemidler: Teraflex, Arthroflex, Arthra, etc.
Mineralmetabolismen stimuleres av legemidler som normaliserer fosfor-kalsiumprosesser. De foretrukne legemidlene er ofte aktive former for vitamin D: Alpha D3 Teva, Oxidevit, Bonviva, etc. Samtidig brukes fosfor-, kalsium- og magnesiumpreparater. Under behandlingen kontrolleres nivået av kalsium og fosfor i blodet eller urinen omtrent en gang hver 20. dag, og det utføres en blodprøve for alkalisk fosfatase.
For å forbedre kroppens bioenergetiske tilstand er det mulig å foreskrive fosfaden, riboksin, lecitin, Elkar, koenzym Q10.
Et omtrentlig behandlingsregime kan se slik ut:
- Kondroprotektor i alderstilpasset dose, tatt sammen med mat og tilstrekkelig mengde vann. Varigheten av én behandlingskur er 3–4 måneder.
- L-prolin i en dose på 500 mg (for barn fra 12 år og voksne) tas en halvtime før måltider, 1-2 ganger daglig, i seks uker. Ved behov kan et aminosyrekompleks i tillegg foreskrives - L-prolin, L-lysin, L-leucin i mengden 10-12 mg/kilogram vekt. Inntaket utføres 1-2 ganger daglig, i to måneder.
- Vitamin- og mineralkomplekspreparater Centrum, Vitrum eller Unicap, i dosering som tar hensyn til alder. Administrasjonsvarighet er 4 uker.
Denne behandlingsregimen er passende hvis en pasient med araknodaktyli klager over problemer med muskel- og skjelettsystemet, og testresultater indikerer økt utskillelse av glykosaminoglykaner i en daglig urintest og reduserte nivåer av frie aminosyrer i blodet.
Som regel blir behandlingen godt mottatt av pasientene, uten noen spesielle bivirkninger. Hvis det oppdages overfølsomhetsreaksjoner, byttes legemidlene ut og behandlingsregimet justeres.
Fysioterapibehandling
Fysioterapeutiske prosedyrer for araknodaktyli foreskrives i henhold til indikasjoner. For eksempel, ved utilstrekkelig osteogenese, for å forbedre helingen av beinbrudd, eller ved tegn på osteoporose, anbefales elektroforese med 5 % kalsiumklorid, 4 % magnesiumsulfat, 2 % kobbersulfat eller 2 % sinksulfat.
Hvis det oppdages vegetative-vaskulære lidelser, brukes 1% natriumkoffeinbenzoat, efedrinhydroklorid eller mesaton.
For å aktivere binyrebarkens funksjon foreskrives medikamentell elektroforese med 1,5 % etimizol og desimeterbehandling i binyreregionen. [ 15 ]
For å stabilisere tonen i blodårene anbefales vannbehandlinger som fremmer vaskulær "gymnastikk". Bad som karbondioksid, furu, kloridhydrogen, hydrogensulfid og radon er utmerkede. Det praktiseres massasjebad, oversvømmelser, kontrastdusjer, skum- og saltbad.
For å forbedre tilstanden til bruskvev brukes magnetisk terapi, induktoterapi, laserterapi og elektroforese med dimetylsulfoksid (Dimexide).
Kirurgisk behandling
Kirurgiske operasjoner for araknodaktyli foreskrives strengt i henhold til indikasjoner. Hvis for eksempel alvorlige hemodynamiske forstyrrelser oppdages mot bakgrunn av prolaps av klaffspissene, utføres massiv aortaaneurisme, klaffutskiftning og skadet segment av aorta. [ 16 ]
Hvis det er uttalte funksjonelle forstyrrelser i luftveiene og kardiovaskulære systemer som oppstår som følge av alvorlig krumning av brystet, utføres thoraxplastikk.
Ved progressivt smertesyndrom forårsaket av alvorlig skoliose på 3–4 grader, er kirurgisk inngrep også indisert. Fra et oftalmologisk synspunkt er absolutte indikasjoner for linsefjerning subluksasjon av linsen komplisert av sekundær glaukom, samt grå stær og degenerative forandringer i netthinnen med høy risiko for linseløsning.
Enhver operasjon på pasienter med araknodaktyli og andre lidelser som påvirker bindevevsstrukturene utføres kun i stadiet med relativ klinisk og biokjemisk lindring. Etter intervensjonen er langvarig medisinsk observasjon og intensiv behandling med legemidler som forbedrer bindevevsmetabolismen obligatorisk.
Forebygging
Araknodaktyli er en sjelden genetisk patologi. Noen ganger oppstår den spontant hos tilsynelatende friske foreldre. Derfor er det svært, svært vanskelig å forebygge sykdommen på forhånd.
Hvis en av de potensielle foreldrene har en belastet familiehistorie – det vil si at det er kjent at noen i familien har lidd av araknodaktyli – bør ektefellene oppsøke en genetiker og gjennomgå en genetisk undersøkelse i planleggingsfasen for et barn. Etter å ha bekreftet graviditeten, utfører legen prenatal diagnostikk, inkludert en ultralydundersøkelse av fosteret og en rekke biokjemiske tester:
- blodprøve fra mor;
- analyse av fostervann;
- prøvetaking av chorionvillus;
- analyse av placentaceller og navlestrengsblod.
Pasienter med araknodaktyli er under medisinsk oppfølging hele livet. Forebyggende tiltak hos pasienter er rettet mot å forhindre komplikasjoner. For dette formålet utføres hjertekirurgisk korreksjon, og medikamentell behandling foreskrives for å eliminere risikoen for trombedannelse. Antibiotikabehandling administreres med jevne mellomrom.
Prognose
Prognosen for livet til pasienter med araknodaktyli bestemmes først og fremst av alvorlighetsgraden av samtidige lidelser - spesielt sykdommer i hjerte- og karsystemet, muskel- og skjelettsystemet og de visuelle organene. Oftest er patologien komplisert av ruptur og disseksjon av aorta. Dette må tas i betraktning, og rettidig hjertekirurgisk korreksjon må utføres, noe som vil bidra til å forlenge pasientens liv og forbedre livskvaliteten.
Hvis diagnosen ble stilt i tide, kan prognosen kalles betinget gunstig hvis legens anbefalinger følges. Medisinsk støtte lindrer araknodaktyliens forløp betydelig, og pasientene har muligheten til å leve et normalt og relativt fullt liv uten å utvikle alvorlige komplikasjoner.