Nye publikasjoner
Nye funn bidrar til en bedre forståelse av årsakene til Rett syndrom
Sist anmeldt: 02.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.

Rett syndrom er en sjelden nevroutviklingsforstyrrelse som det for øyeblikket ikke finnes noen kur eller god behandling for. Det forårsaker alvorlige fysiske og kognitive symptomer, hvorav mange overlapper med autismespekterforstyrrelser.
Rett syndrom er forårsaket av mutasjoner i MECP2-genet, som er høyt uttrykt i hjernen og ser ut til å spille en viktig rolle i å opprettholde nevronenes helse. Genet er lokalisert på X-kromosomet, og syndromet rammer primært jenter. For å utvikle behandlinger for Rett syndrom ønsker forskere å bedre forstå MECP2 og dets funksjoner i hjernen.
Forskere, inkludert Rudolf Jaenisch, medgründer av Whitehead Institute, har studert MECP2 i flere tiår, men mange grunnleggende fakta om genet forble ukjente. Proteinet som er kodet av genet, MECP2, er involvert i genregulering; det binder seg til DNA og påvirker uttrykknivåene til forskjellige andre gener, eller mengden protein de produserer.
Forskerne hadde imidlertid ikke en fullstendig liste over gener som er påvirket av MECP2, og det var ingen enighet om hvordan MECP2 påvirker disse genene.
Tidlige studier av MECP2 antydet at det var en repressor som reduserte uttrykket av målgenene, men forskning av Jaenisch og andre hadde tidligere vist at MECP2 også fungerer som en aktivator, som øker uttrykket av målgenene – og at det kan være en aktivator i utgangspunktet. Også ukjent var MECP2s virkningsmekanisme, eller hva proteinet nøyaktig gjør for å forårsake endringer i genuttrykk.
Teknologiske begrensninger har hindret forskere i å få klarhet i disse spørsmålene. Men Yanish, postdoktor Yi Liu i laboratoriet hans, og Yanishs tidligere laboratoriemedlem Anthony Flamier, nå assisterende professor ved CHU Sainte-Justine forskningssenter ved Université de Montréal, har brukt banebrytende teknikker for å svare på disse gjenværende spørsmålene om MECP2 og få ny innsikt i dens rolle i hjernens helse og sykdom.
Resultatene deres ble publisert i tidsskriftet Neuron, og forskerne opprettet også et nettbasert arkiv for MECP2-dataene sine, MECP2-NeuroAtlas-portalen, som en ressurs for andre forskere.
«Jeg tror denne artikkelen fundamentalt vil endre folks forståelse av hvordan MECP2 forårsaker Rett syndrom. Vi har en helt ny forståelse av mekanismen, og den kan gi nye muligheter for å utvikle behandlinger for sykdommen», sier Janisch, som også er professor i biologi ved MIT.
Dypere forståelse av MECP2 i hjernen
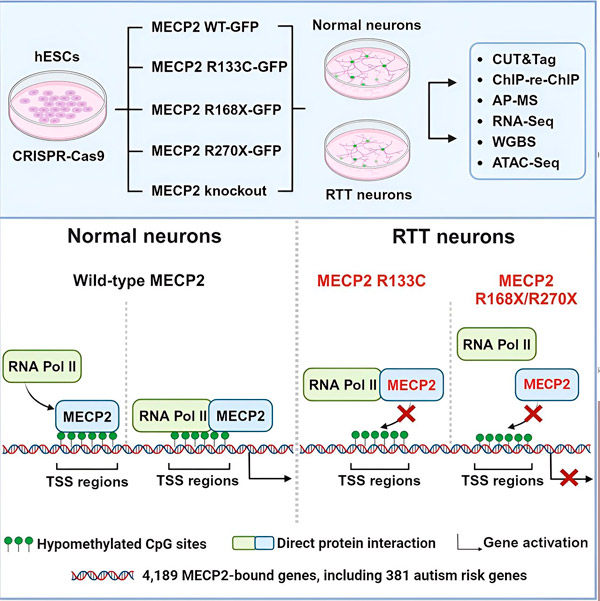
Forskerne laget først et detaljert kart over hvor MECP2 binder seg i menneskelige nevrale gensekvenser, enten innenfor gener eller i regulatoriske regioner av DNA i nærheten av dem. De brukte en metode kalt CUT&Tag, som kan finne proteininteraksjoner med DNA med høy presisjon.
Forskerne fant mer enn 4000 gener assosiert med MECP2. De gjentok kartleggingen sin i nevroner med vanlige MECP2-mutasjoner assosiert med Rett syndrom for å bestemme hvor MECP2 er utarmet i sykdomstilstanden.
Å vite hvilke gener MECP2 binder seg til, gjorde at Liu og Flamier kunne begynne å koble MECP2s mål og hjernens helse. De fant ut at mange av målene er involvert i utviklingen og funksjonen til nevrale aksoner og synapser.
De sammenlignet også listen over MECP2-mål med Simons Foundation Autism Research Initiative (SFARI) database over autismeassosierte gener, og fant at 381 gener i den databasen er MECP2-mål.
Kilde: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Disse funnene kan bidra til å avklare mekanismene bak autismesymptomer ved Rett syndrom og gi et godt utgangspunkt for å undersøke den mulige rollen til MECP2 i autisme.
«Vi har laget det første integrerte kartet over MECP2-epigenomet innen helse og sykdom, og dette kartet kan veilede fremtidig forskning», sier Liu. «Å vite hvilke gener som er mål for MECP2, og hvilke gener som er direkte forstyrret i sykdommen, gir et solid grunnlag for å forstå Rett syndrom og stille spørsmål om genregulering i nevroner.»
Forskerne så også på om MECP2 økte eller reduserte uttrykket av målgenene. I samsvar med historien om at MECP2 ble identifisert av noen som en aktivator og av andre som en repressor, fant Liu og Flamier eksempler der MECP2 spilte begge rollene.
Men selv om MECP2 oftere blir sett på som en repressor, fant Liu og Flamier at det stort sett er en aktivator – noe som bekrefter tidligere funn av Jaenisch og Liu. Et nytt eksperiment viste at MECP2 aktiverer minst 80 % av målene sine, og et annet fant at det aktiverer opptil 88 % av målene sine.
Kartet over målgener som forskerne laget ga ytterligere innsikt i MECP2s rolle som aktivator. De fant ut at gener som MECP2 aktiverer, binder seg vanligvis til en region av DNA oppstrøms for genet kalt transkripsjonsstartstedet.
Dette er stedet der cellulært maskineri starter prosessen med å transkribere et gen til RNA, hvoretter RNA-et blir oversatt til et funksjonelt protein, som er et produkt av genuttrykk. Tilstedeværelsen av MECP2 på transkripsjonsstartstedet, der genuttrykk begynner, er i samsvar med dets rolle som en genaktivator.
Forskerne satte seg deretter fore å finne ut hvilken rolle MECP2 spiller i genaktivering. De så på hvilke molekyler MECP2 binder seg til på dette stedet, i tillegg til DNA, og fant ut at MECP2 samhandler direkte med et proteinkompleks kalt RNA-polymerase II (RNA Pol II). RNA Pol II er en viktig cellulær maskin som transkriberer DNA til RNA. RNA Pol II kan ikke finne gener på egenhånd, så den krever en rekke kofaktorer, eller proteinkollaboratører, for å hjelpe den med å gjøre jobben sin.
Forskerne foreslår at MECP2 fungerer som en slik kofaktor, som hjelper RNA Pol II med å starte transkripsjon i gener der MECP2 binder seg. Strukturanalyse av MECP2 har identifisert deler av molekylet som binder seg til RNA Pol II, og andre eksperimenter har bekreftet at tap av MECP2 reduserer tilstedeværelsen av RNA Pol II på passende transkripsjonsstartsteder, samt uttrykknivåene til målgener.
Dette tyder på at Rett syndrom kan være forårsaket av redusert transkripsjon av gener som er målrettet av MECP2 på grunn av MECP2-mutasjoner som hindrer det i å binde seg til RNA Pol II eller binde seg til DNA. I samsvar med denne ideen er de vanligste MECP2-mutasjonene assosiert med sykdom trunkeringer: mutasjoner der en del av proteinet mangler, noe som kan endre samspillet mellom MECP2 og RNA Pol II.
Forskerne håper at funnene deres ikke bare vil endre vår forståelse av MECP2, men at en dypere og bredere forståelse av hvordan MECP2 påvirker hjernens utvikling og funksjon kan føre til ny innsikt som vil hjelpe personer med Rett syndrom og relaterte lidelser, inkludert autisme.
«Dette prosjektet er et godt eksempel på den samarbeidende naturen til Janisch-laboratoriet», sier Flamier. «Rudolf og jeg hadde et spesifikt problem knyttet til Rett syndrom, og jeg hadde erfaring med CUT&Tag-teknologien, som kunne løse problemet. Gjennom diskusjon innså vi at vi kunne slå oss sammen, og nå har vi et stort arkiv med informasjon om MECP2 og dens koblinger til sykdom.»