Nye publikasjoner
Nøkkelprotein identifisert for å hindre tap av beinmasse ved osteoporose
Sist anmeldt: 02.07.2025

Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
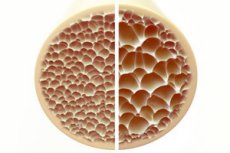
Osteoporose, en tilstand som kjennetegnes av porøse og skjøre bein, utgjør en betydelig trussel mot skjeletthelsen. Bein, som den primære strukturelle støtten til menneskekroppen, gir viktig støtte. Når beinmassen reduseres, svekker det ikke bare denne støtten, men svekker også den generelle funksjonen, noe som fører til redusert livskvalitet.
Etter hvert som forekomsten av osteoporose øker i den aldrende befolkningen, er det en økende belastning på helseressursene for langtidsbehandling. Det er derfor nødvendig å forstå mekanismene som bidrar til utviklingen av osteoporose og å utvikle effektive målrettede behandlinger for å minimere den langsiktige effekten.
Osteoblaster og osteoklaster er to typer celler som spiller en nøkkelrolle i vedlikehold og ombygging av beinvev. Mens osteoblaster er beindannende celler som er ansvarlige for syntese og avsetning av nytt beinvev, er osteoklaster beinnedbrytende celler som er involvert i nedbrytning og fjerning av gammelt eller skadet beinvev.
En økning i andelen osteoklaster fører til bentap ved tilstander som osteoporose, revmatoid artritt (betennelse i leddene) og benmetastaser (kreft som har spredt seg til beinene). Osteoklaster oppstår fra differensieringen av makrofager eller monocytter, som er typer immunceller.
Dermed kan hemming av osteoklastdifferensiering tjene som en terapeutisk strategi for å forhindre bentap. Imidlertid er de nøyaktige molekylære mekanismene som regulerer den komplekse prosessen med beinremodellering fortsatt uklare.
I en ny studie har professor Tadayoshi Hayata, Takuto Konno og Hitomi Murachi fra Tokyo University of Science, sammen med kolleger, forsket på den molekylære reguleringen av osteoklastdifferensiering. Stimulering med reseptoraktivator av kjernefaktor kappa B-ligand (RANKL) induserer differensiering av makrofager til osteoklaster.
I tillegg har signalveier for benmorfogenetisk protein (BMP) og transformerende vekstfaktor (TGF)-β blitt implisert i reguleringen av RANKL-mediert osteoklastdifferensiering. I den nåværende studien hadde forskerne som mål å undersøke rollen til Ctdnep1, en fosfatase (et enzym som fjerner fosfatgrupper) som har vist seg å undertrykke BMP- og TGF-β-signalveier.
Studien er publisert i tidsskriftet Biochemical and Biophysical Research Communications.
Professor Hayata sier: «RANKL fungerer som en «akselerator» for osteoklastdifferensiering. Å kjøre bil krever ikke bare gass, men også bremser. Her fant vi at Ctdnep1 fungerer som en «brems» i osteoklastdifferensiering.»
Forskerne undersøkte først Ctdnep1-ekspresjon i RANKL-behandlede musemakrofager og ubehandlede kontrollceller. De observerte at Ctdnep1-ekspresjon ikke endret seg som respons på RANKL-stimulering. Imidlertid var den lokalisert til cytoplasmaet i granulær form i makrofager og differensiert til osteoklaster, forskjellig fra dens normale perinukleære lokalisering i andre celletyper, noe som indikerer dens cytoplasmatiske funksjon i osteoklastdifferensiering.
Videre resulterte nedregulering av Ctdnep1 (nedregulering av genuttrykk) i en økning i antall osteoklaster positive for tartratresistent syrefosfatase (TRAP), hvor TRAP er en markør for differensierte osteoklaster.
Knockout av Ctdnep1 resulterte i økt uttrykk av viktige differensieringsmarkører, inkludert "Nfatc1", en mastertranskripsjonsfaktor indusert av RANKL for osteoklastdifferensiering. Disse resultatene støtter en "bremsefunksjon" til Ctdnep1, der den negativt regulerer osteoklastdifferensiering. Dessuten resulterte knockout av Ctdnep1 også i økt kalsiumfosfatabsorpsjon, noe som tyder på en undertrykkende rolle for Ctdnep1 i benresorpsjon.
Til slutt, selv om Ctdnep1-knockout ikke endret BMP- og TGF-β-signalering, viste Ctdnep1-defekte celler økte nivåer av fosforylerte (aktiverte) proteiner, som er produkter av RANKL-signalveien. Disse resultatene tyder på at den hemmende effekten av Ctdnep1 på osteoklastdifferensiering kanskje ikke medieres gjennom BMP- og TGF-β-signalering, men gjennom nedregulering av RANKL-signalveien og Nfatc1-proteinnivåer.
Samlet sett gir disse resultatene ny innsikt i osteoklastdifferensieringsprosessen og identifiserer potensielle terapeutiske mål som kan brukes til å utvikle behandlinger for å redusere bentap på grunn av osteoklastoveraktivitet. I tillegg til sykdommer karakterisert av bentap, har Ctdnep1 også blitt identifisert som en årsaksfaktor i medulloblastom, en hjernesvulst hos barn. Forfatterne er optimistiske med tanke på at forskningen deres kan utvides til andre menneskelige sykdommer utover beinmetabolisme.
Professor Hayata konkluderer: «Resultatene våre tyder på at Ctdnep1 er nødvendig for å forhindre overdreven osteoklastogenese. Disse resultatene kan ytterligere utvide vår kunnskap om hvordan fosforylerings-defosforyleringsnettverket kontrollerer osteoklastdifferensiering og kan gi nye terapeutiske strategier for behandling av beinsykdommer forbundet med overdreven osteoklastaktivitet.»